



Microbiome Research Reports







Clara G. de los Reyes-Gavilán , ...Nuria Salazar
, ...Nuria Salazar
, ...Nuria Salazar
Views: Downloads:
Views: Downloads:
Views: Downloads:
Views: Downloads:
Pilar Manrique, ... David Rios-Covian
, ... David Rios-Covian
Views: Downloads:
Joanne M. Donkers, ... Evita van de Steeg
, ... Evita van de Steeg
Views: Downloads:
Views: Downloads:









Data
978
Authors
1579
Reviewers
2021
Published Since
For Reviewers
For Readers
Add your e-mail address to receive forthcoming Issues of this journal:
Themed Collections
Volume 5, Issue 2 (in progress)
Volume 5, Issue 1
Volume 4, Issue 4
Volume 4, Issue 3
Volume 4, Issue 2
Volume 4, Issue 1
Volume 3, Issue 4
Volume 3, Issue 3
Volume 3, Issue 2
Volume 3, Issue 1
Volume 2, Issue 4
Volume 2, Issue 3
Volume 2, Issue 2
Volume 2, Issue 1
Volume 1, Issue 4
Volume 1, Issue 3
Volume 1, Issue 2
Volume 1, Issue 1
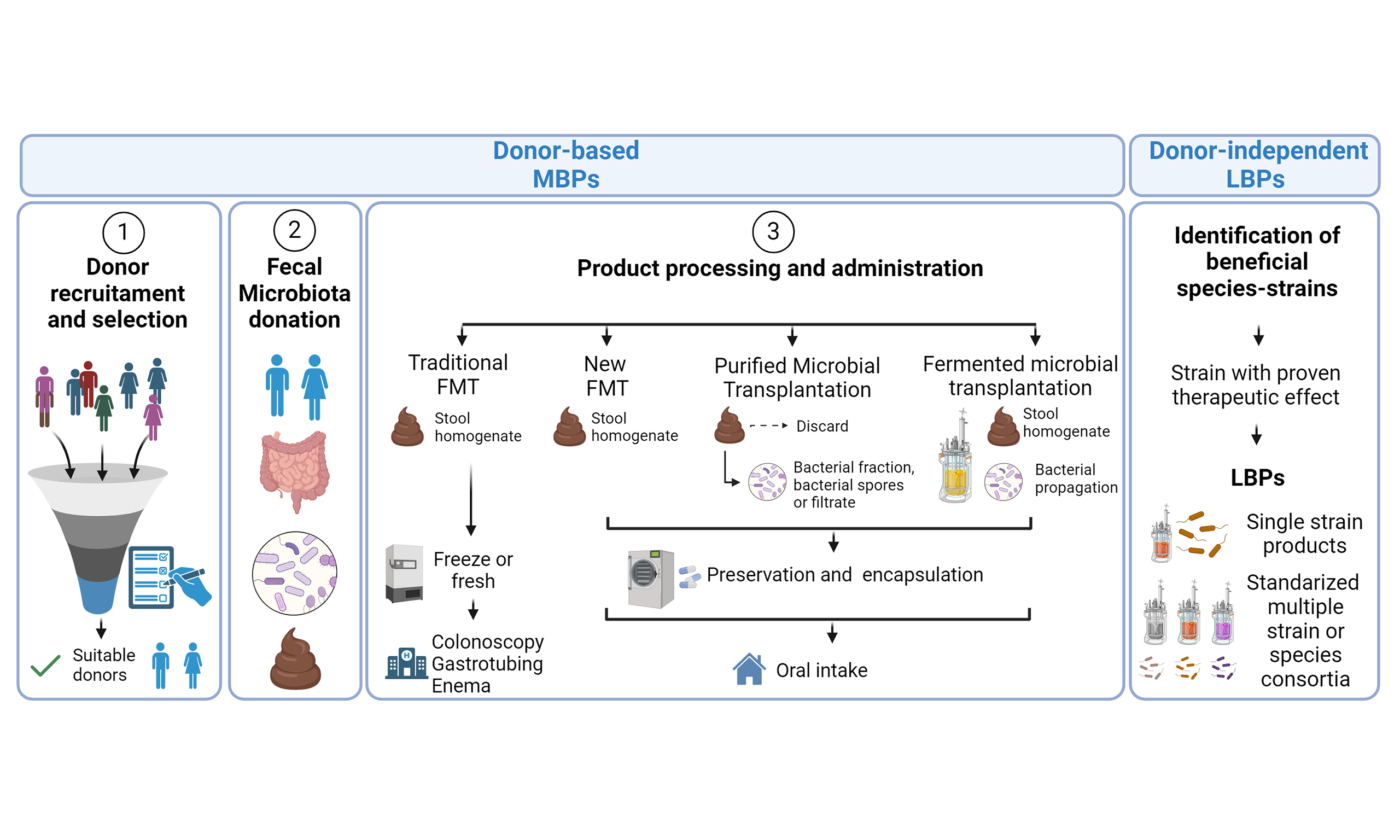
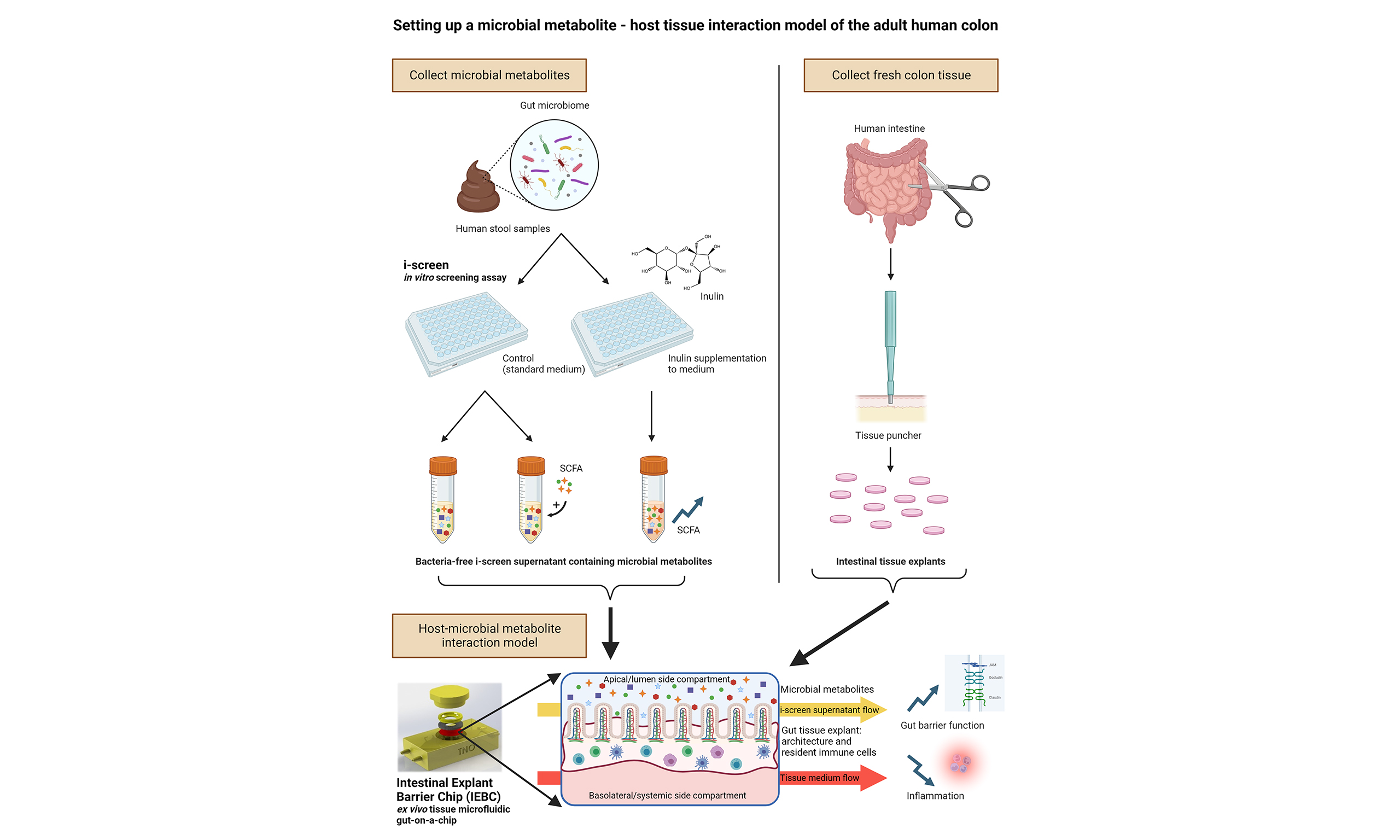
Joanne M. Donkers, ... Evita van de Steeg
, ... Evita van de SteegOpen Access |
Original Article | 21 Feb 2024
Views: | Downloads:

Coming soon.
Volume 5, Issue 2 (in progress)
Volume 5, Issue 1
Volume 4, Issue 4
Volume 4, Issue 3
Volume 4, Issue 2
Volume 4, Issue 1
Volume 3, Issue 4
Volume 3, Issue 3
Volume 3, Issue 2
Volume 3, Issue 1
Volume 2, Issue 4
Volume 2, Issue 3
Volume 2, Issue 2
Volume 2, Issue 1
Volume 1, Issue 4
Volume 1, Issue 3
Volume 1, Issue 2
Volume 1, Issue 1
Data
978
Authors
1579
Reviewers
2021
Published Since




