



Charge/orbital disordered states with smaller volume and higher entropy in transition-metal oxides


Abstract
Some transition-metal oxides such as Ca2RuO4, BiNiO3, and V2OPO4 harbor smaller volume and higher entropy states by role sharing of the spin, charge, and orbital degrees of freedom. Effect of lattice distortions on the various charge/orbital patterns can be analyzed by d-p models with full degeneracy of the transition-metal d and oxygen 2p orbitals. Based on the mean-field analyses on the d-p models for Ca2RuO4, BiNiO3 and V2OPO4, possible mechanisms of negative thermal expansion with charge and orbital degrees of freedom are discussed. In Ca2RuO4 and BiNiO3, orbital and/or charge states are rearranged across their insulator-metal transitions, and the metallic phases with orbital and/or charge fluctuations can be stabilized at high temperatures relative to the insulating phases without them. In V2OPO4, the charge/orbital disordered state can keep relatively smaller volume due to orbital-dependent hybridization in the face-sharing VO6 octahedron chain.
Keywords
INTRODUCTION
Transition-metal oxides exhibit surprisingly rich electrical, magnetic, and structural properties due to the correlated d electrons[1,2]. The d orbitals with five-fold degeneracy in the atomic limit are split into three-fold degenerate t2g (xy, yz, and zx) and two-fold degenerate eg (3z2-r2 and x2-y2) orbitals under the cubic ligand field. When the number of d electrons per transition-metal site is integer, the d electrons can be localized with the strong on-site electron-electron interaction (Mott insulators). A typical phase diagram of such Mott insulators is illustrated in Figure 1A. For example, pyrite-type NiS2 exhibits a pressure-induced phase transition from a Mott insulator to a paramagnetic metal[1,2]. The paramagnetic metallic phase with itinerant d electrons has smaller volume than the Mott insulating phase with localized d electrons. In the Mott insulating phase, the localized spins tend to order antiferromagnetically (sometimes ferromagnetically) unless strong frustration effect sets in due to lattice geometry or other degrees of freedom. In NiS2 with a face-centered cubic lattice, the Ni2+ ion (d8 configuration with S = 1) does not have orbital degrees of freedom, and the S = 1 spins are antiferromagnetically ordered at low temperatures. In the paramagnetic insulating phase, the disordered spins can provide entropy of kBlog3 per Ni site. Since the symmetry of the paramagnetic insulating phase is the same as that of the paramagnetic metallic phase, their phase boundary is terminated at the critical point. In such a case without orbital degrees of freedom, the paramagnetic insulating phase with relatively large volume is stabilized at high temperatures due to the spin entropy

Figure 1. (A) Schematic phase diagram of NiS2 as functions of temperature T and pressure P. (B) Schematic phase diagram of Ca2RuO4. PI, AFI, PM, and AFM represent paramagnetic insulating, antiferromagnetic insulating, paramagnetic metallic, and antiferromagnetic metallic phases. (C) Energy per unit cell calculated by mean-filed approximation for the layered perovskite-type d-p model with d4 as a function of Jahn-Teller distortion (the ratio between the compressed/elongated apical Ru-O bond length and the in-plane Ru-O bond length. F and AF represent ferromagnetic and antiferromagnetic states. With the compression (elongation), the xy (yz or zx) orbitals are doubly occupied.
When the charge/orbital orderings are associated with magnetic ordering and electron-electron interaction, the charge/orbital ordered states at low temperatures tend to have relatively large volume due to the localized d electrons with weak hybridization between neighboring d electrons. When the charge/orbital ordering is driven by formation of spin singlet bonds due to electron-lattice interaction (such as CuIr2S4 and MgTi2O4), the charge/orbital ordered state at low temperature has smaller volume. In the former case, since the exchange interaction between localized spins depends on the charge and orbital arrangement, the magnetic ordering temperature is usually lower than the charge and/or orbital ordering temperature. There are several types of lattice distortions to stabilize their charge/orbital order: Jahn-Teller distortion for
METHODS
The spin-charge-orbital order/disorder transitions are analyzed by mean-field calculations on d-p models in which the d-d Coulomb interaction is expressed by Kanamori parameters u, u′ and j with u′ = u-2j[10,11]. The transfer integrals between the transition-metal d and oxygen 2p orbitals are given by Slater-Koster parameters (pds) and (pdp) with the ratio (pdp)/(pds) = -0.45. Following the previous studies on the d-p model for 3d and 4d transition-metal oxides[11,12], u′ and j are set to 3.0 and 0.5 eV for Ca2RuO4, 7.0 and
RESULTS AND DISCUSSION
Layered perovskite-type Ca2RuO4 exhibits an insulator-metal transition around 350 K and an antiferromagnetic transition at 110 K [Figure 1B][16-19]. RuO6 octahedra share their corners forming a square lattice of Ru in the ab plane of the layered perovskite. The Ru 4d t2g orbitals accommodate four electrons (two holes) in the low spin configuration. In the low (high) temperature phase of Ca2RuO4, the RuO6 octahedron is compressed (elongated) along the c-axis or the z-axis and the xy (yz/zx) orbital is stabilized relative to the yz/zx (xy) orbital among the three t2g orbitals. As shown in Figure 1C, the orbital occupancy change due to the compression and elongation of the octahedron can be described by mean-filed calculations although the spin and orbital fluctuations are not considered. The compressed case has no orbital degrees of freedom. In the elongated case, the doubly degenerate yz/zx orbitals accommodate one hole per Ru site which may provide orbital entropy of kBlog2 and spin entropy of kBlog3 in the localized limit of S = 1. If the Ru 4d yz/zx electrons are fully itinerant, the spin and orbital entropy should be reduced. However, the strong electronic correlation in Ca2RuO4 suggests that the orbital contribution may remain even in the metallic phase.
The spin entropy can be reduced even in the paramagnetic insulating phase since the space-time fluctuations of spin and orbital are restricted by Ru 4d spin-orbit interaction[3]. While the RuO6 octahedron is strongly compressed in the antiferromagnetic insulating phase, it is almost regular in the paramagnetic insulating phase. When the energy splitting between the xy and yz/zx orbitals becomes comparable or smaller than the spin-orbit interaction, the three t2g orbitals are mixed to form the complex orbitals of
and
where ↑ and ↓ indicate spin up and down. Under the strong spin-orbit interaction,
Perovskite-type RNiO3 (R = rare earth) becomes insulating at low temperatures due to charge disproportionation of 2Ni3+→Ni2++Ni4+. The metallic phase has smaller volume and is thus stabilized by pressure, as schematically shown in Figure 2A[20-22]. The charge disproportionation is stabilized by the breathing type distortion of the NiO6 octahedra, as illustrated in Figure 2B. The effect of the breathing type distortion can be studied by mean-filed calculations on the perovskite-type d-p model. The results

Figure 2. (A) Schematic phase diagram for RNiO3 as functions of temperature T and pressure P. PI, AFI, PM, and CD represent paramagnetic insulating, antiferromagnetic insulating, paramagnetic metallic, and charge disproportionated phases. (B) Distortions of the NiO6 octahedron and the electronic configurations for the charge disproportionated Ni2+ and Ni4+ sites without oxygen 2p holes. The yellow and red circles indicate oxygen and transition metal ions, respectively. (C) Energy per unit cell for ferromagnetic and antiferromagnetic (A, G, and C type) states calculated by mean-filed approximation for the perovskite-type d-p model with d7 as a function of breathing distortion (the ratio between the long and average Ni-O bond length). Without the distortion, each site takes the low spin d7 state with S = 1/2. With the distortion, the expanded site becomes d8 (S = 1) and the compressed site becomes d6 (S = 0). (D) Electronic configuration of d8L. (E) Electronic configuration of d8L2.
Since the phase diagram of RNiO3 resembles that of Ca2RuO4, RNiO3 itself may exhibit NTE. However, the Ni-O bond length is considerably shortened in the Ni4+O6 octahedron due to the low-spin d8L2 configuration which is hybridized with the low-spin d7L and d6 configurations. The volume of the charge-disproportionated insulating state is only slightly larger than that of the metallic state, and the NTE effect would be limited. Instead, Azuma et al. have established that a sister material BiNiO3 exhibits one of the best NTE performances[6]. In the insulating phase of BiNiO3, the oxygen hole of Ni3+(d8L) is taken by Bi and the Ni valence becomes +2[18]. As a result, the valence state of Bi3+0.5Bi5+0.5Ni2+O3 is realized where the configuration of Bi5+ is s2L2 rather than s0. Since the Ni2+-O bond length is longer than the Ni4+-O one, the volume of the insulating BiNiO3 is larger than that of the charge disproportionated RNiO3. Also, the Bi 6s2 lone pair introduces additional lattice distortions to BiNiO3. As expected, pressure or chemical substitution (such as Bi1-xLaxNiO3) induces a valence transition from Bi3+0.5Bi5+0.5Ni2+O3 to Bi3+Ni3+O3 with substantial volume collapse[6]. This transition can be viewed as oxygen hole transfer from the Bi5+ site to the Ni3+ site, and this picture is indeed supported by the recent ab initio calculations[27].
In the NTE BiNiO3, the Ni2+-O-Ni2+ superexchange interaction is enhanced due to the relatively small but positive charge-transfer energy (about 4 eV), and the magnetic order (or short-range order) can survive near the Ni-to-Bi charge transfer transition under pressure or with chemical substitution. Therefore, the spin entropy of the insulating Bi3+0.5Bi5+0.5Ni2+O3 tends to be suppressed. The metallic Bi3+Ni3+O3 can be viewed as a charge-disordered metallic state of the charge-disproportionated RNiO3. Theoretically, the phase diagram has been studied using mean field calculations on a negative U Hubbard model[13]. In the charge-disordered metallic state, oxygen holes are dissociated from the Ni and Bi sites. Most probably, in the metallic phase of Bi3+Ni3+O3 and Bi3+1-xLa3+xNi3+O3, the d8L state of Ni3+ is disassembled into d8 at the Ni site and L. While the oxygen hole L is responsible for the metallicity, the localized d8 state can provide spin entropy of kBlog3 per Ni site. This picture is too simplified because the hybridization between the d8L and d7 configurations and the band formation are neglected. In the metallic phase, the spin entropy of the localized limit should be reduced due to the d8L-d7 hybridization and the band formation. However, substantial spin and charge entropy can remain in the metallic state where the bandwidth is reduced to the order of 0.1 eV by the strong electron-electron and electron-lattice interaction. In this sense, the spin degrees of freedom of Ni 3d8 state and the charge degrees of freedom of oxygen hole are correlated in BiNiO3 providing the NTE[6,28].
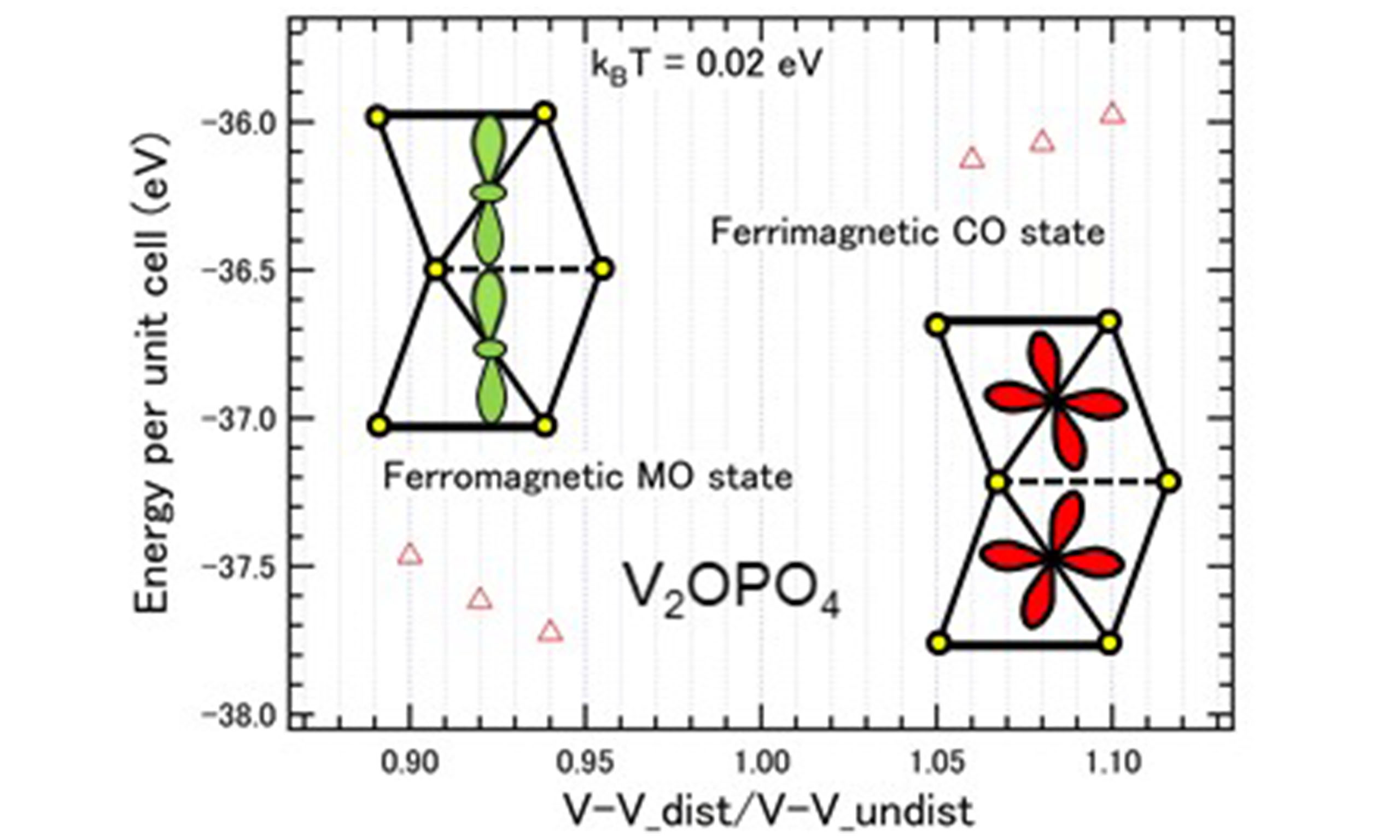
V2OPO4 consists of face-sharing and edge-sharing VO6 octahedra and is supposed to have a body-centered tetragonal unit cell[7,29]. However, it undergoes V2+/V3+ charge ordering at 605 K with monoclinic lattice distortion. The face-sharing V2+ and V3+ sites form chains along the [110] direction of the monoclinic lattice which corresponds to the a-axis of the tetragonal lattice of the high-temperature phase. The corner-sharing V3+ sites are connected approximately along the [001] direction of the monoclinic lattice which is inclined by ~30 degrees relative to the c-axis of the tetragonal lattice. The face-sharing V2+ and V3+ chain along the [110] direction is illustrated in Figure 3A. The x, y, and z axes are along the V-O bonds. In the present work, a d-p model with a face-sharing octahedron chain is employed. The ferrimagnetic charge-ordered (CO) state is stable only when the V-V bond is elongated or the VO6 octahedron is elongated along the chain. The spin difference between the neighboring V sites is about 0.6. When the octahedron is close to the regular shape, there is no stable solution. Interestingly, another mean-field solution appears when the V-V bond is substantially compressed. In this solution, the spin difference between the neighboring V sites is as small as 0.1, indicating formation of the molecular orbital (MO) as illustrated in Figure 3B. The calculated energies are plotted as a function of compression/elongation along the V-V bond [Figure 3C].

Figure 3. (A) Ferrimagnetic CO state viewed along the face-sharing VO6 chain and from the side of the chain. The yellow circles indicate oxygen ions. The x, y, and z axes are along the V-O bonds. (B) Ferromagnetic MO state viewed from the side of the chain. The a1g orbitals form the MO. (C) Energy per unit cell (open triangles) calculated by mean-filed approximation for the face-sharing octahedron d-p model with d2.5 as a function of compression/elongation along the V-V bond (ratio between the compressed/elongated V-V bond length and the original V-V bond length).
In the real system, the VO6 octahedron of V3+ is compressed along the z-axis in the CO phase. Therefore, the xy orbital is lower in energy than the yz/zx orbitals. Considering the Hund coupling, one of the yz/zx orbitals is occupied at the V3+ site as shown in Figure 3A. Such orbital order is consistent with polarization dependence of V 2p X-ray absorption spectrum[9]; consequently, the orbital fluctuation is quenched.
CONCLUSIONS
Based on the mean-field calculations on the d-p models, the relationship between the charge/orbital states and the lattice distortion has been discussed for Ca2RuO4, RNiO3, and V2OPO4. The changes of the charge/orbital states by the lattice distortion suggest their disordered states harbor relatively high entropy due to charge/orbital fluctuations. Across the insulator-metal transition in Ca2RuO4, the RuO6 octahedron is elongated and the yz/zx orbital fluctuation becomes relevant in the metallic phase. The yz/zx orbital fluctuation in the metallic phase is intimately related to the orbital-dependent band renormalization in the metallic phase of Ca2-xSrxRuO4[32]. Above the transition temperature of Ca2RuO4, the strongly renormalized Ru 4d electrons (bandwidth ~0.1 eV) are almost incoherent and exhibit localized character. The insulator-metal transition in BiNiO3 is accompanied by the oxygen hole transfer from Bi to Ni. In V2OPO4, the charge/orbital disordered state has relatively small volume due to the a1g-a1g bond in the face-sharing octahedra while the spin, charge, and orbital fluctuations of the egπ electrons provide higher entropy than the charge/orbital ordered state. In Ca2RuO4 and V2OPO4, the orbital-dependent fluctuations of the t2g electrons play key roles in realizing the NTE behaviors. While the orbital order/disorder is coupled with the Jahn-Teller distortion in the corner-sharing octahedra of Ca2RuO4, it is governed by the metal-metal dimerization in the face-sharing octahedra of V2OPO4. Such t2g electron systems with orbital ordering can be candidates for new NTE materials. Orbital and/or charge states are rearranged across their insulator-metal transitions of Ca2RuO4 and BiNiO3, and the metallic phases with orbital and/or charge fluctuations can be stabilized at high temperatures relative to the insulating phases without them. The negative charge-transfer energy and the oxygen 2p holes are responsible for the charge-transfer mechanism and provide the charge fluctuations in the high-temperature phase of BiNiO3. In Ca2RuO4, the Jahn-Teller distortion gradually develops below the transition temperature due to the spin-orbit coupling[3]. The spin-orbit interaction would be important in the extremely wide temperature range of NTE in Ca2RuO4.
DECLARATIONS
Acknowledgments
The author would like to thank Prof. G. A. Sawatzky, Prof. L. H. Tjeng, Prof. S. Nakatsuji, Prof. Y. Maeno, Prof. D. I. Khomskii, Prof. M. Azuma, Prof. J. P. Attfield, and the members of their research groups for long-term collaborations on the target materials of this article.
Authors’ contributions
The author contributed solely to the article.
Availability of data and materials
Data supporting the findings of this article are available from the author upon reasonable request.
Financial support and sponsorship
This work was supported by the JSPS KAKENHI Grant (No. JP22H01172).
Conflicts of interest
The author declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Imada, M.; Fujimori, A.; Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 1998, 70, 1039-263.
3. Mizokawa, T.; Tjeng, L. H.; Sawatzky, G. A.; et al. Spin-orbit coupling in the Mott insulator Ca2RuO4. Phys. Rev. Lett. 2001, 87, 077202.
4. Takenaka, K.; Inoue, N.; Mizuno, Y.; et al. Extended operating temperature window of giant negative thermal expansion in Sn-doped Ca2RuO4. Appl. Phys. Lett. 2018, 113, 071902.
5. Alonso, J. A.; García-Muñoz, J. L.; Fernández-Díaz, M. T.; Aranda, M. A. G.; Martínez-Lope, M. J.; Casais, M. T. Charge disproportionation in RNiO3 perovskites: simultaneous metal-insulator and structural transition in YNiO3. Phys. Rev. Lett. 1999, 82, 3871-4.
6. Azuma, M.; Chen, W. T.; Seki, H.; et al. Colossal negative thermal expansion in BiNiO3 induced by intermetallic charge transfer. Nat. Commun. 2011, 2, 347.
7. Pachoud, E.; Cumby, J.; Lithgow, C. T.; Attfield, J. P. Charge order and negative thermal expansion in V2OPO4. J. Am. Chem. Soc. 2018, 140, 636-41.
8. Murota, K.; Pachoud, E.; Attfield, J. P.; et al. Charge correlation in V2OPO4 probed by hard x-ray photoemission spectroscopy. Phys. Rev. B. 2020, 101, 235159.
9. Murota, K.; Pachoud, E.; Attfield, J. P.; et al. Vanadium 3d charge and orbital states in V2OPO4 probed by x-ray absorption spectroscopy. Phys. Rev. B. 2020, 101, 245106.
10. Mizokawa, T.; Fujimori, A. Unrestricted hartree-fock study of transition-metal oxides: spin and orbital ordering in perovskite-type lattice. Phys. Rev. B. Condens. Matter. 1995, 51, 12880-3.
11. Mizokawa, T.; Fujimori, A. Electronic structure and orbital ordering in perovskite-type 3d transition-metal oxides studied by Hartree-Fock band-structure calculations. Phys. Rev. B. Condens. Matter. 1996, 54, 5368-80.
12. Kurokawa, M.; Mizokawa, T. Orbital state and metal-insulator transition in Ca2-xSrxRuO4 studied by model Hartree-Fock calculations. Phys. Rev. B. 2002, 66, 024434.
13. Naka, M.; Seo, H.; Motome, Y. Theory of valence transition in BiNiO3. Phys. Rev. Lett. 2016, 116, 056402.
14. Mizokawa, T.; Khomskii, D. I.; Sawatzky, G. A. Spin and charge ordering in self-doped Mott insulators. Phys. Rev. B. 2000, 61, 11263-6.
15. Yoshino, T.; Okawa, M.; Kajita, T.; et al. Unusual valence state and metal-insulator transition in BaV10O15 probed by hard x-ray photoemission spectroscopy. Phys. Rev. B. 2017, 95, 075151.
16. Mizokawa, T.; Tjeng, L. H.; Lin, H.; et al. Orbital state and metal-insulator transition in Ca2-xSrxRuO4 (x=0.0 and 0.09) studied by x-ray absorption spectroscopy. Phys. Rev. B. 2004, 69, 132410.
17. Nakamura, F.; Goko, T.; Ito, M.; et al. From mott insulator to ferromagnetic metal: a pressure study of Ca2RuO4. Phys. Rev. B. 2002, 65, 220402.
18. Steffens, P.; Friedt, O.; Alireza, P.; et al. High-pressure diffraction studies on Ca2RuO4. Phys. Rev. B. 2005, 72, 094104.
19. Keen, H. D. J.; Julian, S. R.; Hermann, A. Ab initio study of pressure-induced structural and electronic phase transitions in Ca2RuO4. Phys. Rev. B. 2021, 104, 085143.
20. Torrance, J. B.; Lacorre, P.; Nazzal, A. I.; Ansaldo, E. J.; Niedermayer, C. Systematic study of insulator-metal transitions in perovskites RNiO3 (R=Pr,Nd,Sm,Eu) due to closing of charge-transfer gap. Phys. Rev. B. Condens. Matter. 1992, 45, 8209-12.
21. Zhou, J. S.; Goodenough, J. B.; Dabrowski, B. Pressure-induced non-Fermi-liquid behavior of PrNiO3. Phys. Rev. Lett. 2005, 94, 226602.
22. Cheng, J.; Zhou, J.; Goodenough, J. B.; Alonso, J. A.; Martinez-lope, M. J. Pressure dependence of metal-insulator transition in perovskites RNiO3 (R=Eu , Y, Lu). Phys. Rev. B. 2010, 82, 085107.
23. Mizokawa, T.; Fujimori, A.; Arima, T.; Tokura, Y.; Mori, N.; Akimitsu, J. Electronic structure of PrNiO3 studied by photoemission and x-ray-absorption spectroscopy: band gap and orbital ordering. Phys. Rev. B. Condens. Matter. 1995, 52, 13865-73.
24. Johnston, S.; Mukherjee, A.; Elfimov, I.; Berciu, M.; Sawatzky, G. A. Charge disproportionation without charge transfer in the rare-earth-element nickelates as a possible mechanism for the metal-insulator transition. Phys. Rev. Lett. 2014, 112, 106404.
25. Green, R. J.; Haverkort, M. W.; Sawatzky, G. A. Bond disproportionation and dynamical charge fluctuations in the perovskite rare-earth nickelates. Phys. Rev. B. 2016, 94.
26. Wang, L.; Yang, Z.; Bowden, M. E.; et al. Hole-trapping-induced stabilization of Ni4 + in SrNiO3/LaFeO3 superlattices. Adv. Mater. 2020, 32, e2005003.
27. Paul, A.; Mukherjee, A.; Dasgupta, I.; Paramekanti, A.; Saha-Dasgupta, T. Hybridization-switching induced mott transition in ABO3 perovskites. Phys. Rev. Lett. 2019, 122, 016404.
28. Nishikubo, T.; Sakai, Y.; Oka, K.; et al. Optimized negative thermal expansion induced by gradual intermetallic charge transfer in
29. Pachoud, E.; Cumby, J.; Wright, J.; Raguz, B.; Glaum, R.; Attfield, J. P. Electronic origin of negative thermal expansion in V2OPO4. Chem. Commun. 2020, 56, 6523-6.
30. Chang, C.; Koethe, T.; Hu, Z.; et al. c-axis dimer and its electronic breakup: the insulator-to-metal transition in Ti2O3. Phys. Rev. X. 2018, 8, 021004.
31. Miyoshino, T.; Takegami, D.; Meléndez-Sans, A.; et al. Intra c-axis dimer hybridization and mixed valency in Mg-doped Ti2O3. Phys. Rev. B. 2023, 107, 115145.
Cite This Article

How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright















Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].