


Machine learning descriptors for crystal materials: applications in Ni-rich layered cathode and lithium anode materials for high-energy-density lithium batteries


Abstract
Lithium batteries have revolutionized energy storage with their high energy density and long lifespan, but challenges such as energy density limitations, safety, and cost still need to be addressed. Crystalline materials, including Ni-rich cathodes and lithium anodes, play pivotal roles in the performance of high-energy-density lithium batteries. Understanding the micro-scale behavior and degradation mechanisms of these materials is crucial for improving macro-scale battery performance. Simulation methods, particularly machine learning (ML) techniques, have become indispensable tools in elucidating these intricate processes because of great efficiency and acceptable accuracy. ML methods depend on descriptors, which bridge the gap between crystal structures and input matrices of models. These descriptors encode essential atomic-level details in crystal structures, enabling predictions of material properties and behaviors relevant to lithium batteries. This paper reviews and discusses the diverse array of descriptors employed in the simulation of crystalline materials for lithium batteries with high energy density. Case studies highlight the effectiveness of different descriptors in simulating cathode behaviors such as Li/Ni disordering, screening of stable LiNi0.8Co0.1Mn0.1O2 (NMC811) configurations, and lithium deposition behaviors at the anode interface. The discussed descriptors can also be applied to other crystalline cathode, anode, and electrolyte materials in lithium batteries and advance the development of lithium batteries with superior energy density.

Keywords
INTRODUCTION
The increasing lifespan demand for electric vehicles necessitates improvements in the energy density of lithium batteries. To enhance energy density, battery material performance is crucial. Ni-rich cathode materials and lithium anode materials are often studied for improving battery energy density[1-5]. In recent years, machine learning (ML) has become widely used in the development of Ni-rich cathode and lithium anode materials[5-7], with effective crystal descriptors for representing these crystalline materials enhancing ML efficiency and enabling more precise predictions[8]. As illustrated in Figure 1, crystal structures and properties are digitalized into crystal descriptors, including explicit crystal structure descriptors, graph descriptors, and encoding methods to further optimize these descriptors. These descriptors are employed in ML tasks such as property prediction[9-12], atomistic simulations[13-15], and crystal generation[16-18], and the outputs can then be used for large-scale materials discovery and development. This review focuses on the recent advances of ML crystal descriptors and the applications of these crystal descriptors in Ni-rich cathode and lithium anode materials, accelerating exploration on materials for high-energy-density lithium batteries.

Figure 1. Crystal descriptors for ML-driven development of lithium battery materials. Different descriptors, which digitally describe crystal structures, possess distinct strengths that help accelerate diverse ML tasks including property prediction, atomistic simulation, and crystal generation. The results from ML tasks can be used as new input structures and properties for further model training and prediction. ML: Machine-learning.
Among the crystal descriptors used in ML studies, traditional explicit descriptors were the first to be proposed and utilized. Since then, various descriptors have been organized into toolkits for convenient use, including Magpie[19], Pymatgen[20], Matminer[21], and DScribe[22,23], among others. These explicit descriptors typically include the basic parameters of a supercell that represent the crystal structure. Standard parameters, such as lattice constants, atomic and bonding properties, symmetry operations, and macroscale properties such as charge-discharge data can be easily extracted and have been used for decades in materials science. In previous studies, these types of descriptors have been applied in property prediction tasks, including energy, modulus, and other focused characteristics[5,24-28]. For example, Guo et al. extracted explicit descriptors from partial charging voltage curves and used ML models, including XGBoost, to predict the battery impedance spectrum[29]. However, the limitations of explicit crystal descriptors become apparent when more complex properties, related to local structures or interactions, need to be predicted or when the models must generalize across a wide range of materials with various crystal structures or key characteristics.
Traditional explicit crystal structure descriptors, while foundational and easily obtained, often fall short in the accuracy and transferability required by modern ML models for predicting material properties, especially in complex atomistic simulations. To address these limitations, researchers have developed complex descriptors that provide a more detailed representation of crystal structures. One such advancement is the development of atom-centered descriptors. From the smooth overlap of atomic positions (SOAP) descriptor[30], many-body tensor representation (MBTR) descriptor[31], to more complex moment tensor potential (MTP) descriptor[32,33] and atomic cluster expansion (ACE) descriptor[34], which satisfied the requirement of completeness, these descriptors collect atom-centered information and incorporate them together to describe the entire crystal structure. Another major advancement is the use of graph-based descriptors[35-41]. In this approach, the crystal structure is naturally represented as a graph where atoms are nodes, and the bonds between them are edges. Graph-based crystal descriptors allow for a more detailed and flexible description of the crystal structure, and can be easily augmented with advanced physical and chemical properties[36,38,42-44].
Atom-centered and graph-based descriptors well represent detailed information in crystal structures, but complex encodings are necessary for special tasks. Graph neural networks (GNNs) have emerged as powerful tools for processing these atom-centered and graph-based descriptors. By using message passing among descriptor components and incorporating equivariant constraints, GNNs can effectively capture the complex relationships of the graph with fewer parameters[45], making them highly suitable for tasks such as property prediction and ML force fields (MLFF)[14,46,47]. For instance, GNNs have been used to predict electronic properties, stability, and reactivity of materials with remarkable accuracy[5,8,36,43,48-50]. Another innovative approach that is gaining traction is the use of graph-based descriptors combined with encoding techniques such as Transformer networks. Originally developed for natural language processing, transformers have shown great potential in capturing long-range dependencies and complex patterns, which are often present in molecular and crystal structures[51]. While GNNs excel at capturing local interactions within a crystal, transformers can process more global, long-range interactions. This combination can lead to more accurate and comprehensive predictions of material properties and enables special ML tasks such as crystal generation. Although Transformer networks have seen various applications in molecular structures[4,47], their use in processing crystal structures is still emerging[17,52].
In the following sections, a comprehensive overview of crystal descriptors and their applications in lithium batteries with high energy density is provided. In the section on explicit crystal descriptors, we introduce the widely used traditional explicit crystal descriptors along with a brief discussion of their applications. The section on complex crystal descriptors covers advancements in atom-centered and graph-based descriptors, which offer clearer crystal-structural descriptions and higher accuracy for predictions. To meet the demand of more specific and complicated material simulation tasks, the section on encoding and embedding enhanced crystal descriptors explores the development of embedding methods to enhance the capability of descriptors, particularly for MLFF and crystal generation tasks. Eventually, in the section on applications of crystal descriptors in high-energy-density lithium batteries, we discussed the detailed applications of crystal descriptors in the ML-assisted development of Ni-rich cathode materials and lithium anode materials for high-energy-density lithium batteries.
EXPLICIT CRYSTAL DESCRIPTORS
Traditional explicit crystal descriptors, which are fundamental for converting crystal structures into inputs for ML models, are often extracted directly from these structures and include parameters such as lattice constants, atomic numbers, bond lengths, and more. A typical collection of these descriptors is provided by Matminer[19,21], an open-source Python library specifically designed to aid researchers in creating and manipulating descriptors for materials science. Developed by the Materials Project team, Matminer [Figure 2A] is integrated with widely-used databases such as Materials Project (MP)[53,54] and Open Quantum Materials Database (OQMD)[55], and provides a wide array of tools that enable efficient data retrieval, descriptor calculation, and data analysis, with an extensive collection of pre-built descriptors, which encompass structural, compositional, and electronic properties of materials. Descriptor functions implemented in Matminer include, but are not limited to, compositional descriptors (stoichiometric attributes, element property statistics), structural descriptors (radial distribution functions, bond length statistics), and electronic descriptors (density of states, band structure features). These descriptors can be readily computed for a given material dataset, providing a rich set of features that can be used in subsequent ML tasks. Various research on materials for lithium batteries has been conducted by Matminer. For instance, a study was conducted by Kim et al. to predict the average voltage and volume change of doped Ni-rich cathode materials [LiNi0.85Dx’D’’(0.15-x)O2] for lithium-ion batteries[56]. The descriptors used in this model were chemical and structural descriptors generated based on Matminer, and additional features including space group number, degree of de-lithiation, and gravimetric capacity. Another research by Min et al. uses traditional descriptors [13 synthesis parameters, inductively coupled plasma mass spectrometry (ICP-MS) and X-ray diffraction (XRD) features] combined with AdaBoost-ERT model to predict initial capacity, cycle life and the amount of residual Li of Ni-rich cathode materials (LiNixCoyMn1-x-yO2, x > 0.85) from an experimental dataset with 330 data points, and further optimize the synthesis procedures[11]. Besides, the descriptors in Matminer were combined with SHapley Additive exPlanations (SHAP) analysis[57,58] to understand the black-box-like ML and further explain Li/Ni disordering mechanism in LiNiO2 and LiNi0.8Co0.1Mn0.1O2 (NMC811) cathode materials[59,60].

Figure 2. Explicit crystal structure descriptors and universal descriptor packages. (A) Matminer, including Magpie descriptors representing composition, valence electrons and other basic parameters of crystal materials. This figure is quoted with permission[21], Copyright 2018, Elsevier; (B) Automatminer, a tool for automatically creating complete ML pipelines for materials science, including automatic featurization with Matminer, feature reduction, and an AutoML backend. Put in a materials dataset and get out a machine that predicts materials properties. This figure is quoted with permission[61], Copyright 2020, Springer Nature. ML: Machine learning; AutoML: automated machine learning.
Based on Matminer, Automatminer [Figure 2B] is developed as an automated ML (AutoML) library tailored for materials science applications[61]. Automatminer aims to simplify the development of ML models by automating the processes of feature engineering, model selection, and hyperparameter tuning. Automatminer employs a modular architecture that integrates seamlessly with Matminer for data acquisition, feature generation, and further model determination. It automatically selects and computes the most relevant descriptors from Matminer, ensuring that the feature set is both comprehensive and tailored to the specific prediction task at hand. This automated feature engineering process not only saves time but also improves the likelihood of identifying the most predictive features. In addition to feature engineering, Automatminer provides robust algorithms for model selection and optimization, enabling Automatminer to identify the best-performing ML models for a given dataset. The automated hyperparameter tuning further enhances model performance by systematically exploring the parameter space to find the optimal settings. Additionally, Automatminer provides a platform called Matbench[62] for benchmarking ML model performance on material design tasks, benefiting the reproduction and improvement of existing ML-assisted materials research.
COMPLEX CRYSTAL DESCRIPTORS
Explicit crystal descriptors are easy to prepare and use, but they often lack comprehensive details about crystal structures, making them insufficiently precise or transferable across different systems. To address this limitation, improvements have been made to crystal descriptors, leading to the development of more complex representations. One of the advancements is atom-centered symmetry descriptors collected in the DScribe library[22,23] [Figure 3], including complex materials descriptors such as SOAP[30] in Figure 3A, atom-centered symmetry functions (ACSF)[63] in Figure 3B, MBTR[31] in Figure 3C, and Valle-Oganov descriptor[64] in Figure 3D. Similarly, MTP is proposed by Shapeev et al. and demonstrates high accuracy among different MLFFs[13,32,33]. These atom-centered descriptors transform the local crystal environment into different basis encoded functions, and all atom-centered functions are aggregated to fully represent a whole material structure (both crystalline and molecular). For example, the generative adversarial network (GAN)-based ML model Crystalor[65] utilizes the SOAP descriptor to encode crystal structures and further generate relaxed Li-M-N-O cathode structures from unrelaxed inputs.

Figure 3. Atom-centered symmetry descriptors implemented in the DScribe library. (A) DScribe is a Python and C library for converting materials into descriptors for ML research, reprinted with permission[22], Copyright 2019, Elsevier; (B) SOAP descriptor, reprinted with permission[22], Copyright 2019, Elsevier; (C) ACSF descriptor, reprinted with permission[22], Copyright 2019, Elsevier; (D) MBTR descriptor; (E) Valle-Oganov descriptor, reprinted with permission[23], Copyright 2019, Elsevier and Copyright 2023, AIP Publishing. SOAP: Smooth overlap of atomic positions; ACSF: atom-centered symmetry functions; MBTR: many-body tensor representation.
Another type of complex crystal descriptor is a graph-based crystal descriptor. Among these, property-labeled materials fragments (PLMF)[66] was proposed early and worked as a fragmented graph descriptor composed of nodes with various combinations of atom properties and edges with atomic connectivity determined by Voronoi polyhedral in crystal structures. As shown in Figure 4A, PLMF reduces the complexity of crystal graph descriptors by decomposing them into smaller, representative subgraphs with no more than three consecutive, non-repetitive edges. The crystal-wide properties, including lattice parameters, density, and space groups, are concatenated to form the final descriptor. The PLMF descriptors describe the crystal structures in more detail and have been proven efficient in predicting key crystal properties (band gap energy, modulus, and so on) when combined with gradient boosted decision tree (GBDT) model. They have also inspired other graph-based universal crystal descriptors. Furthermore, SchNet [Figure 4B][15] improves the graph-based crystal descriptor by introducing GNN to convert the whole crystal graphs (instead of fragmented graphs) into fixed-length vectors. Specifically, filter-generating convolutional networks are employed to include the rotational invariance and periodic boundary conditions thus better describing the whole crystal environment. As a result, SchNet is able to learn chemically plausible embeddings of atom types and atomic environments with lattice constraints. Apart from property prediction, the crystal descriptor proposed in SchNet is further improved to predict potential energy surfaces and is applied in ML molecular dynamics calculations[13].

Figure 4. Graph-based descriptors. (A) PLMF[66], adapted with permission[66], Copyright 2017, Springer Nature; (B) SchNet, a deep learning architecture for crystals and molecules[15]; (C) graph descriptor of CGCNN[43] reprinted with permission[43], Copyright 2018, American Physical Society; (D) MEGNet with state parameters included in crystal graph[37], adapted with permission[37], Copyright 2019, American Chemical Society. PLMF: Property-labeled materials fragments; CGCNN: crystal graph convolutional neural networks; MEGNet: MatErials graph network.
Another widely used graph-based crystal descriptor is implemented in crystal graph convolutional neural networks (CGCNN)[43], designed to predict material properties by representing crystal structures as graphs [Figure 4C]. In CGCNN, atoms are represented as nodes, and bonds are edges. The network applies convolutional operations on these graphs to learn the underlying physics and chemistry. The key features include graph representation, which naturally captures the periodicity and connectivity of crystal structures, versatility in predicting a wide range of material properties, and a data-driven approach that benefits from extensive training on large datasets. CGCNN has been employed to predict high-voltage and high-conductivity cathode materials for lithium-ion batteries[67]. Due to its concise framework and strong description ability, CGCNN has been a basic and benchmarking model in ML research on battery materials, tasks including predicting average voltage[9], specific capacity[9,10], and oxidation potential[42] of electrode materials. To further incorporate local environment information in graph descriptors, MEGNet[37,48] was developed by Chen et al. and exhibited superior performance on materials property predictions. The descriptor in MEGNet includes global state variables [Figure 4D], which can represent external conditions such as temperature or pressure, in graph convolution process and make it universal for both crystalline materials and molecules. In detail, the global state vectors are updated along with atom vectors and bond vectors using message passing mechanism proposed in message passing neural networks (MPNN)[68,69], and pooling technique is employed to ensure invariance in crystal and molecule structures. With these designs, MEGNet demonstrated superior performance in various property prediction tasks including band gap, energy, formation energy, and so on. He et al. use MEGNet to predict energy order and obviously reduce the number of Li-Mn-rich layered cathode calculations and further accelerate the simulation of Li-Mn-rich cathode’s evolutions under different current densities[70]. Since MEGNet has demonstrated aptitude in material property domain, it often serves as baseline model for benchmarking when developing new descriptors or new models on tasks including cathode oxidation potential prediction[42,44,71]. A comparison of the property prediction capabilities of these graph-based descriptors, combined with different ML models, has been conducted and is presented in MatBench[61,62].
ENCODING AND EMBEDDING ENHANCED CRYSTAL DESCRIPTORS
Crystal descriptors serve as the initial input for ML models when processing crystalline materials in high-energy-density lithium batteries, and they perform well in simpler tasks such as property prediction and statistical analysis. However, more complicated ML tasks arise in the simulations of high-energy-density lithium batteries, necessitating specialized encoding and embedding on descriptors to meet these task requirements.
In recent years, ML has been extensively applied in fitting MLFF and accelerating molecular dynamics simulations. One of the most commonly used MLFF model is DeepMD, with its descriptors implemented in the DeepMD-kit[72-74] software package [Figure 5A]. In this descriptor, the atomic positions in crystal structure are used to generate the local environment matrix, which is then trained in the embedding layer to generate final descriptor matrix for the fitting neural network. The DeepMD model had been further extended to a universal atomic model, DPA[46,47], with unified descriptors and can be applied in diverse tasks including MLFF, property prediction, and structure generation when combined with different output layers. Besides, MatErials 3-body graph network (M3GNet) improves MEGNet by incorporating 3-body interactions and lattice matrix to obtain force and stress tensors[36] [Figure 5B]. Crystal Hamiltonian graph neural network (CHGNet) incorporates charge and magnetic moment information to consider electron interactions and orbital occupancy, thus suitable for transition metal systems [Figure 5C], and has been applied to simulate the phase transformation of Li0.5MnO2 and LixFePO4 phase diagram[75]. Another widely-used graph tensor MLFF model, MACE[14,76], includes four-body messages instead of traditional two-body messages, thus obviously reducing the message passing cost of MPNNs [Figure 5D]. Besides, equivariant GNN is used to process distance vectors between atoms and conduct the message passing among crystal graphs, different from the distance scalars used in SchNet and MEGNet. Thus, MACE combines the advantages of high-order message passing and equivariant MPNNs. MACE potential has been applied in various fields of battery materials including probing Jahn-Teller distortions in LiNiO2 cathode materials[77]. These interatomic potentials enhance aforementioned crystal descriptors by well-designed network structure to include more physical information, thus keeping ab-initio-level accuracy while offering opportunity to perform traditional molecular dynamics calculations, and benefiting large-scale simulations with high accuracy.

Figure 5. Encoding and embedding enhanced crystal descriptors for MLFF. (A) Crystal descriptor proposed in DeepMD[72,74], which is further expanded in DPA universal model; (B) Crystal descriptor enhanced in M3GNet, as an update to MEGNet[36]; (C) Crystal descriptor enhanced in CHGNet, quoted with permission[75], Copyright 2023, Springer Nature; (D) Crystal descriptor enhanced in MACE[14,76]. MLFF: Machine learning force field; M3GNet: MatErials 3-body graph network; CHGNet: crystal Hamiltonian graph neural network.
Apart from MLFF, crystal generation is another specific task related to the inverse design of crystal materials[78]. To achieve reasonable generation results, specialized encoding of descriptors and neural network construction is essential. The variational autoencoder (VAE) is widely used in processing and generating new data with similar characteristics, and has been combined with basic graph descriptors for materials development. For example, crystal diffusion VAE (CDVAE) is a framework that uses message-passing algorithms to model atomic interactions in materials [Figure 6A][16], using SE(3) equivariant GNNs adapted with periodicity (PGNNs) for both the encoder and decoder of VAE. Each atom in a material structure passes messages to its neighbors, allowing the network to learn the dependencies and interactions effectively. The key features of CDVAE include the dynamic exchange of information between atoms through message passing, adaptability to different types of materials and properties, and computational efficiency, making it suitable for large-scale simulations. Compared to CDVAE, Fourier-transformed crystal properties (FTCP) [Figure 6B] employ a similar VAE framework, but further introduce embedding on atom position and lattice matrix in the encoding-decoding process for crystal generation[79]. Besides, attention mechanism is also an efficient way of embedding materials data[17,42,46,52]. The MOFormer model [Figure 6C], based on the Transformer encoder, processes a tokenized MOFid input, which includes SMILES tokenization for secondary building units (SBUs) and separate tokenization for topology and catenation[52]. This tokenized sequence, following the BERT structure with tokens and a fixed length of 512, handles molecular representations. To enhance the performance of describing classical metal-organic framework (MOF) structures, this model especially addresses its limitation in capturing geometric information and introducing a self-supervised learning (SSL) paradigm using CGCNN[43], which processes 3D atomic structures of MOFs. Inspired by the Crystal Twins framework, this approach employs the Barlow Twins loss function to align the embeddings from both MOFormer and CGCNN. After SSL pretraining, the encoder weights are fine-tuned for improved property predictions. This combined SSL pretraining significantly enhances the performance of both models across datasets. Another diffusion model, MatterGen
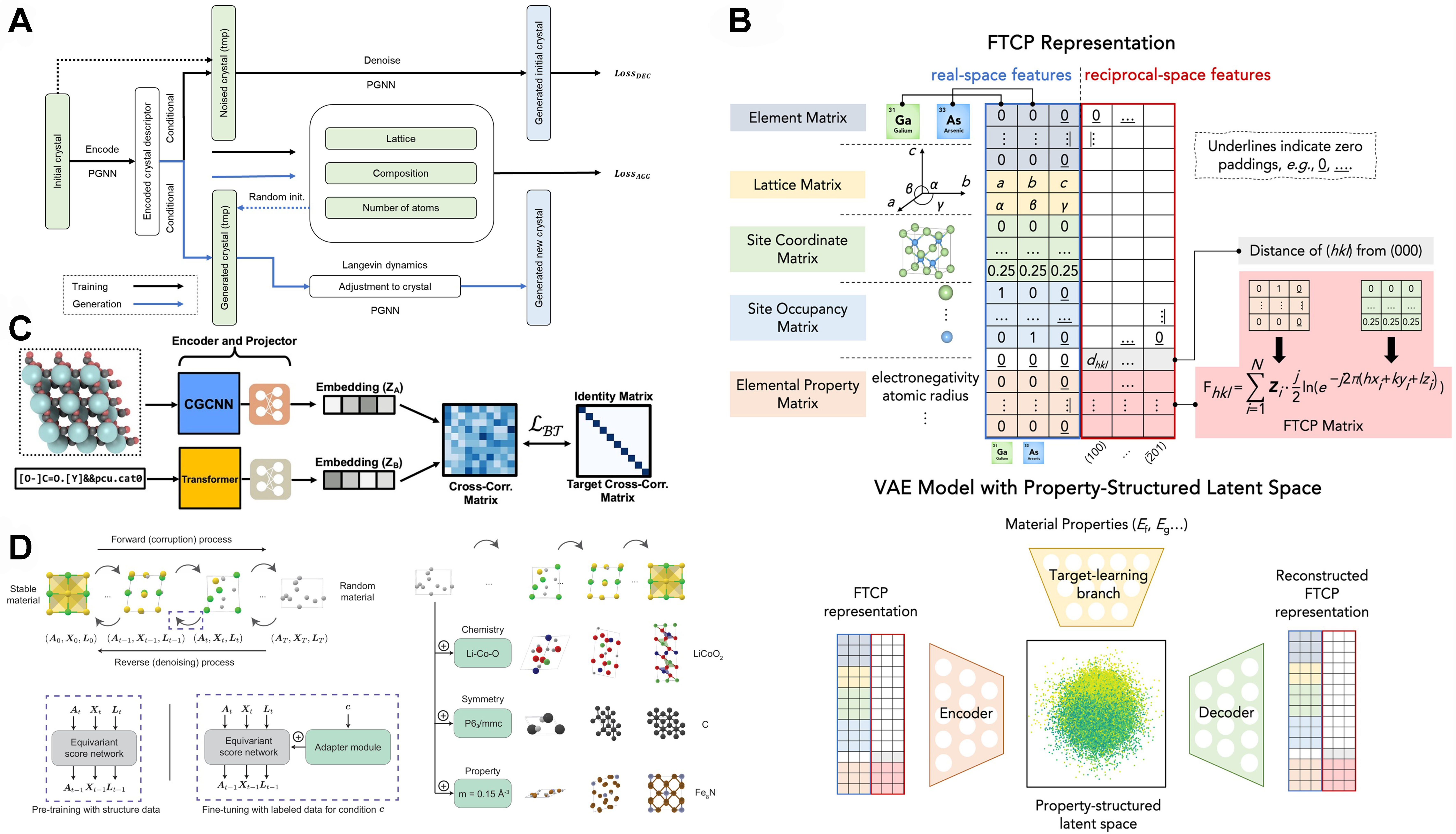
Figure 6. Encoding and embedding enhanced crystal descriptors for crystal generation tasks. (A) CDVAE model for crystalline material generation[16], using conditional denoising technique to generate new structures with requirements; (B) FTCP model using VAE framework for generating novel crystalline structures. Reprinted with permission[79], Copyright 2022, CellPress; (C) MOFormer, encoding framework integrating self-supervised learning framework with CGCNN and transformer. Reprinted with permission[52], Copyright 2023, American Chemical Society; (D) MatterGen, a diffusion model for crystal generation. Reprinted with permission from Zeni et al.[80]. CDVAE: Crystal diffusion variational autoencoder; FTCP: Fourier-transformed crystal properties; VAE: variational autoencoder; CGCNN: crystal graph convolutional neural networks.
APPLICATIONS OF CRYSTAL DESCRIPTORS IN HIGH-ENERGY-DENSITY LITHIUM BATTERIES
The application of these crystal descriptors and ML models has significant implications of the development of lithium battery materials. The use of appropriate descriptors facilitates more accurate predictions of key properties in high-energy-density battery materials, such as Li/Ni disordering behavior[81-83], structural stability[81,83-91], thermal stability[82,92-97], lithium deposition[98-103], ionic conductivity[98,104-107], and other important properties[108-112]. Several aforementioned cases would be discussed in detail here. By accelerating the discovery of new materials with optimal properties, these descriptors can contribute to the development of improved lithium batteries. Moreover, the ability to rapidly screen large libraries of materials using ML models can significantly reduce the time and cost associated with experimental testing.
One of the most focused applications of crystal descriptors in lithium battery materials is property prediction. For Ni-rich cathode materials, the Li/Ni exchange defect is easy to form and severally influences the reversibility of lithium transportation in the batteries[113,114]. As illustrated in Figure 7A, explicit crystal descriptors can be utilized in interpretable ML, and help explain the control mechanisms of doping effect on Li/Ni exchange defect[60]. Different doping elements and diverse local configurations for the antisite Ni ion manifest coupled effects on the Ni-rich layered cathode materials. To understand the complex effects of different doping elements and local configurations of antisite Ni ions, researchers used a random forest (RF) model combined with SHAP analysis to identify important features influencing the Li/Ni exchange defect formation energy (Ef). The initial descriptor set, consisting of 257 features generated by the Magpie–Deml–Pymatgen featurizer in Matminer[21], was insufficient for accurate predictions (R2 = 0.385). Recognizing this limitation, the researchers conducted a detailed analysis to capture the unique physical and chemical characteristics of Li/Ni exchanged layered cathodes, resulting in a customized 100-feature descriptor. Combining these custom features with those from Matminer improves the model performance (R2 = 0.566). Ultimately, the 18 most relevant features were determined by correlation analysis, which reduced prediction error, and this distinct descriptor enhances the ability of the RF model to understand Li/Ni exchange configurations effectively. With this descriptor and RF-SHAP method, it is possible to efficiently identify important properties that influence the Li/Ni exchange defect, and enable effective control of Li/Ni exchange defect. Similarly, Kim et al. used descriptors generated by Matminer and self-defined descriptors to predict the average voltage and volume change of doped Ni-rich cathode materials [LiNi0.85Dx’D’’(0.15-x)O2], finding doping elements including Y, B, and Ga exhibiting excellence in improving performance[56] [Figure 7B].

Figure 7. Crystal descriptors applied in predicting properties of Ni-rich cathode materials. (A) Explicit crystal descriptor-assisted interpretable ML on Li/Ni exchange in Ni-rich cathode materials, quoted with permission[60], Copyright 2024, American Chemical Society; (B) Explicit crystal descriptor-assisted screening on doping elements to obtain better voltage and volume performance, quoted with permission[56], Copyright 2023, Elsevier; (C) Graph-based crystal descriptor and combined ML to accelerate stable NMC811 structure screening and stability analysis (part of ongoing research from our group and currently unpublished, reprinted with permission from the authors). ML: Machine learning; NMC811: LiNi0.8Co0.1Mn0.1O2.
For more accurate property prediction, graph-based descriptors might be more suitable. In one of our recent ongoing studies, the MEGNet descriptor was utilized to rapidly predict the equilibrium structure and the energy of NMC811 cathode materials and, finally, identify the stable NMC811 structures. In this study, the ML-assisted energy prediction model, MEGNet, augmented by transfer learning (TL), demonstrates robust performance in energy prediction tasks despite a limited training dataset comprising several hundred data points [Figure 7C]. This capability stems from leveraging domain knowledge from inorganic crystals, enabling rapid adaptation and accurate predictions with minimal target domain input data. Additionally, the ML-driven structure relaxation model, Crystalor[65], effectively serves as a surrogate to costly density functional theory (DFT) calculations combined with descriptors from MEGNet, enhancing the consistency and reliability of predicting configuration stability for virtual configurations deviating from equilibrium states. Integrating these ML workflows significantly streamlines the identification of the top ten stable configurations from a vast chemical coordination environment encompassing over 500,000 potential initial configurations for NMC811. The predictions of these ML models consistently align with DFT results, validating their efficacy in discerning stable configurations based on both energy and voltage profiles. Moreover, the ML workflow serves as a potent tool for exploring the expansive latent space of complex NMC811 configurations, offering insights into configuration stability studies and revealing preferences for uniformly distributed Ni, Co, and Mn atoms within stable structures.
Another application of crystal descriptors in high-energy-density lithium battery materials is fitting MLFF to accelerate large-scale molecular dynamics simulations under ab initio accuracy. By utilizing embedded crystal descriptors, MLFF merges the accuracy of ab initio calculations with the efficiency of classical force fields and has been successfully applied to several systems. As illustrated in Figure 8, Lai et al. used MLFF to systematically study the lithium metal anode and its interface[115-119]. Firstly, The Li surface deep potential (Li-SP) model was developed using descriptors of DeepMD framework[72,73] [Figure 8A, upside], and was employed to investigate deposition behaviors and mechanisms in lithium metal batteries. In experimental research, high current densities favor inhomogeneous deposition and subsequent Li-dendrite growth attributed to electric field tip effects or uneven electrode surfaces[120-122]. Using Li-SP simulations, this phenomenon was replicated to study dendrite growth mechanisms and explore suppression strategies [Figure 8B]. Results showed two hemispherical dendrites merging into a larger structure upon contact, accompanied by cavity formation that gradually disappeared during fusion, illustrating a “bulk self-healing” process distinct from surface self-healing. Further simulations of Li crystal growth with cuboid cavities demonstrated gradual shrinkage and transformation into amorphous Li, enhancing fluidity and promoting cavity closure. Experimental validation through Li foil self-healing experiments at room temperature confirmed the feasibility and universality of bulk self-healing in lithium metal, contrary to previous simulation findings. These findings highlight the potential for mitigating dendrite formation in lithium-ion batteries through controlled deposition conditions. To further study the mechanism of Li deposition on Cu substrates for anode-free lithium batteries, the lithium-copper neural network interatomic potential (LiCu-NNIP) model was trained based on DeepMD [Figure 8C]. The Cu (100), Cu (110), and Cu (111) surface structures were constructed to investigate Li deposition characteristics[118]. The Cu (100) and Cu (111) surfaces exhibited flat Li-Cu interfaces with ordered Li layers, suggesting surface-limited Li diffusion without alloying. Conversely, on the Cu (110) surface, alloying occurred as evidenced by Cu-Li atom exchange, resulting in a disordered interface. Quantitative analysis using the surface similarity analysis (SSA) method compared Li monolayer structures with standard Li crystal surfaces, revealing that initial Li layers significantly influenced subsequent layer structures, with the first two layers being particularly influential. Additionally, simulations employing an MTP model explored the role of external pressure in facilitating dendrite-free Li formation during Li crystal growth with hole defects at 300 K[116] [Figure 8A, downside]. Results indicated that external pressure accelerated the closure of small holes, while large holes persisted without pressure but gradually shrank and disappeared with its application [Figure 8D]. The presence of amorphous Li atoms near the holes enhanced self-healing by increasing fluidity compared to crystalline Li. These findings underscore the role of external pressure in promoting Li self-healing, particularly for challenging hole defects, facilitating their gradual closure and disappearance.

Figure 8. Crystal descriptors applied in MLFF for atomic simulation of lithium anode materials. (A) Schema of DeepMD and MTP potential, adapted with permission[115,116], Copyright 2022, Wiley-VCH and Copyright 2023, Elsevier; (B) Lithium dendrite self-healing simulation pairs with experimental observations, adapted with permission[117], Copyright 2022, Wiley-VCH; (C) Li-Cu surface simulated by MLFF, adapted with permission[118], Copyright 2022, Wiley-VCH; (D) Large-scale molecular dynamics simulations of Li dendrite growth under different external pressures were performed with a MTP ML force field. External pressure was found to promote the process of Li self-healing. This figure is quoted with permission[116], Copyright 2023, Elsevier. MTP: Moment tensor potential; MLFF: machine learning force fields; ML: machine learning.
As for crystal generation, which is a crucial step of inverse design, embedded crystal descriptors also found applications. Inverse design has been consistently discussed in the development of novel materials with targeted properties[123], which starts from the specific properties and then finds materials with targeted properties. When an existing material database cannot satisfy the requirements on properties, generating and screening novel structures becomes essential. Inverse design has been applied in generating materials with specific symmetry [Figure 9A], magnetic, electronic and mechanical properties [Figure 9B][80]. Among these properties, modulus is crucial to obtaining dendrite-resistant solid-state electrolytes and artificial solid-electrolyte interphase materials[124,125]. Similar approaches can be extended to other key properties, such as lithium conductivity and thermal stability, thereby enhancing the inverse design of lithium battery materials, including solid-state electrolytes and coatings for electrodes.

Figure 9. Crystal descriptors applied in crystal generation task. Generating crystals with target (A) symmetry, (B) magnetic, electronic, and mechanical properties. Reprinted with permission from Zeni et al.[80].
From fundamental research to applied engineering, different types of crystal descriptors with diverse augmentations are suitable for different kinds of tasks in advancing the development of high-energy-density lithium battery materials. As summarized and compared in Table 1, the wide range and versatility of crystal descriptors allows systematic comparisons across different crystalline materials, thereby elucidating structure-property relationships and facilitating materials design for high-energy-density lithium batteries. To be more detailed, in the study of Li/Ni disordering, explicit crystal descriptors enable the identification of diverse atomic configurations and their effects on structure stability and electrochemical properties. For stable configuration screening of NMC811, the graph-based crystal descriptors facilitate the identification of optimal atomic distributions that enhance structural integrity and mitigate degradation during charge-discharge cycles. In the realm of lithium deposition behaviors, equivariant-network-enhanced crystal descriptors help construct MLFF and explain the mechanisms governing dendrite formation, surface roughness, and electrolyte interactions, crucial for developing strategies to enhance battery safety and longevity. As for crystal generation and inverse design, generative-model-enhanced crystal descriptors make it possible to create reasonable virtual crystal structures, which is beneficial to designing novel lithium conductors as solid-state electrolytes.
Comparisons among different kinds of ML crystal descriptors discussed in this review
| Type | Typical implementation | Description | Applications | |
| Explicit crystal descriptors | Electronic/chemical descriptors | Matminer[21], Automatminer[61], self-defined | Descriptors relevant to the electronic structure of crystal: ionization energy, magnetic moment, charge, etc. | Small datasets, property prediction, interpretable tasks |
| Structural descriptors | Matminer[21], Automatminer[61], self-defined | Descriptors relevant to the structure of crystal: lattice parameters, cell volume, density, bond information, etc. | Small datasets, property prediction, interpretable tasks | |
| Complex crystal descriptors | Atom-centered descriptors | DScribe[22,23]: SOAP[30], ACSF[63], MBTR[31], Valle-Oganov descriptor[64] | Descriptors use atom-centered information and basis-functions to represent crystal structure | Large datasets, accurate property prediction |
| Graph-based descriptors | PLMF[66], SchNet[15], CGCNN[43], MEGNet[37,48] | Descriptors use fragmented or whole crystal graphs to represent crystal structure | Large datasets, accurate property prediction | |
| Encoding and embedding enhanced crystal descriptors | High-order graph message passing, equivariant constraints | DeepMD[72,73], M3GNet[36], CHGNet[75], MACE[14,76] | Combining high-order graph message pass and/or physical equivariant limitations with descriptors to learn crystal information with fewer parameters compared to learn from scratch | Large datasets, MLFF |
| Generative models | CDVAE[16], FTCP[79], MOFormer[52], MatterGen[80] | Combining VAE, attention mechanism, and/or diffusion model to meet the requirement of generative tasks | Large datasets, crystal generation | |
CONCLUSION AND OUTLOOK
Crystal descriptors have been extensively researched as ML model inputs in the study and development of materials for Ni-rich cathode materials and lithium anode materials in high-energy-density lithium batteries. These descriptors play a pivotal role in digitalizing the structural and electronic properties of crystalline materials, facilitating predictions and understanding of their performance and behavior in battery applications. Moreover, the crystal descriptors serve as crucial inputs of ML models for further property prediction, computation acceleration, structure generation and other potential applications in advancing materials science. In this article, we provided a comprehensive overview of the types of crystal descriptors and their corresponding applications in advancing the material research for high-energy-density lithium batteries. Explicit crystal descriptors, which are straightforward to develop and apply, have been widely used in the property prediction and mechanism elucidation of various Ni-rich cathode materials. More advanced descriptors, including atom-centered and graph-based descriptors, offer a more detailed representation of crystal structures, enabling more precise prediction tasks. Additionally, some encoding and embedding methods have been developed to suit crystal descriptors for complicated tasks including MLFF and crystal generation, thus enabling ML-assisted molecular dynamics simulations and inverse design.
While the advancements in crystal descriptors and ML models are promising, several challenges persist in their widespread application. One of the primary challenges is the need for large, high-quality datasets to effectively train these models. For complex properties such as ionic conductivity, collecting and curating such datasets is time-consuming and expensive, often requiring extensive experimental or simulation data. The models themselves must also be robust and generalizable to be useful in real-world scenarios, where the materials or conditions may differ from those seen in the training datasets. Another significant hurdle is the interpretability of these ML models. Although advanced models such as GNNs with equivariant constraints, transformer, and generative models deliver accurate predictions, the underlying reasons for their predictions often remain opaque. While specialized neural network frameworks are proposed continuously, the “black box” nature makes it difficult to understand how specific crystal descriptors lead to certain material property predictions, posing a barrier to gaining deeper scientific understandings. Developing methods for interpreting and explaining these predictions is essential, with efforts aimed at making ML models more transparent and providing clearer insights into their physical and chemical mechanisms. Moreover, integrating different types of descriptors and models adds another layer of complexity. Combining the strengths of various approaches - such as graph-based descriptors with diffusion models - requires careful design and optimization. Striking a balance between crystal descriptor complexity and computational efficiency is crucial to making these tools viable for practical applications in enhancing performance of high-energy-density battery materials.
DECLARATIONS
Authors’ contributions
Contributed to the conception and design of the study: Zhang R, Ye Y, Zheng J
Performed data acquisition: Zhang R, Rong F, Lai G, Wu G
Revised the manuscript: Ye Y, Lai G, Zheng J
Provided supervision and acquired funding: Zheng J
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (No.52272180) and the Shenzhen Science and Technology Research Grant (No.20220810123501001).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. Zheng Y, Yao Y, Ou J, et al. A review of composite solid-state electrolytes for lithium batteries: fundamentals, key materials and advanced structures. Chem Soc Rev 2020;49:8790-839.
2. Jiang Z, Li J, Yang Y, et al. Machine-learning-revealed statistics of the particle-carbon/binder detachment in lithium-ion battery cathodes. Nat Commun 2020;11:2310.
3. Wang C, Aoyagi K, Aykol M, Mueller T. Ionic conduction through reaction products at the electrolyte-electrode interface in all-solid-state Li+ batteries. ACS Appl Mater Interfaces 2020;12:55510-9.
4. Gao YC, Yao N, Chen X, Yu L, Zhang R, Zhang Q. Data-driven insight into the reductive stability of ion-solvent complexes in lithium battery electrolytes. J Am Chem Soc 2023;145:23764-70.
5. Chen X, Liu X, Shen X, Zhang Q. Applying machine learning to rechargeable batteries: from the microscale to the macroscale. Angew Chem Int Ed Engl 2021;60:24354-66.
6. Lombardo T, Duquesnoy M, El-Bouysidy H, et al. Artificial intelligence applied to battery research: hype or reality? Chem Rev 2022;122:10899-969.
7. Chen C, Zuo Y, Ye W, Li X, Deng Z, Ong SP. A critical review of machine learning of energy materials. Adv Energy Mater 2020;10:1903242.
8. Li S, Liu Y, Chen D, Jiang Y, Nie Z, Pan F. Encoding the atomic structure for machine learning in materials science. WIREs Comput Mol Sci 2022;12:e1558.
9. Wang S, Ji Y, Liu J, et al. Integrating crystal structure and numerical data for predictive models of lithium-ion battery materials: a modified crystal graph convolutional neural networks approach. J Energy Storage 2024;80:110220.
10. Zhang Z, Wang Y, Li S, Li S, Chen M. Interpretable machine learning prediction of voltage and specific capacity for electrode materials. Adv Theor Simul 2024;7:2400227.
11. Min K, Choi B, Park K, Cho E. Machine learning assisted optimization of electrochemical properties for Ni-rich cathode materials. Sci Rep 2018;8:15778.
12. Saal JE, Kirklin S, Aykol M, Meredig B, Wolverton C. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 2013;65:1501-9.
13. Han T, Li J, Liu L, Li F, Wang L. Accuracy evaluation of different machine learning force field features. New J Phys 2023;25:093007.
14. Batatia I, Benner P, Chiang Y, et al. A foundation model for atomistic materials chemistry. arXiv. [Preprint.] Mar 1, 2024 [accessed on 2024 Oct 21]. Available from: https://doi.org/10.48550/arXiv.2401.00096.
15. Schütt KT, Sauceda HE, Kindermans PJ, Tkatchenko A, Müller KR. SchNet - a deep learning architecture for molecules and materials. J Chem Phys 2018;148:241722.
16. Xie T, Fu X, Ganea OE, Barzilay R, Jaakkola T. Crystal diffusion variational autoencoder for periodic material generation. arXiv. [Preprint.] Mar 14, 2022 [accessed on 2024 Oct 21]. Available from: https://doi.org/10.48550/arXiv.2110.06197.
17. Cao Z, Luo X, Lv J, Wang L. Space group informed transformer for crystalline materials generation. arXiv. [Preprint.] Aug 16, 2024 [accessed on 2024 Oct 21]. Available from: https://doi.org/10.48550/arXiv.2403.15734.
18. Gebauer NWA, Gastegger M, Schütt KT. Symmetry-adapted generation of 3d point sets for the targeted discovery of molecules. arXiv. [Preprint.] Jan 9, 2020 [accessed on 2024 Oct 21]. Available from: https://doi.org/10.48550/arXiv.1906.00957.
19. Ward L, Agrawal A, Choudhary A, Wolverton C. A general-purpose machine learning framework for predicting properties of inorganic materials. npj Comput Mater 2016;2:BFnpjcompumats201628.
20. Ong SP, Richards WD, Jain A, et al. Python materials genomics (pymatgen): a robust, open-source python library for materials analysis. Comput Mater Sci 2013;68:314-9.
21. Ward L, Dunn A, Faghaninia A, et al. Matminer: an open source toolkit for materials data mining. Comput Mater Sci 2018;152:60-9.
22. Himanen L, Jäger MO, Morooka EV, et al. DScribe: library of descriptors for machine learning in materials science. Comput Phys Commun 2020;247:106949.
23. Laakso J, Himanen L, Homm H, et al. Updates to the DScribe library: new descriptors and derivatives. J Chem Phys 2023;158:234802.
24. Deml AM, O’Hayre R, Wolverton C, Stevanović V. Predicting density functional theory total energies and enthalpies of formation of metal-nonmetal compounds by linear regression. Phys Rev B 2016;93:085142.
25. Jha D, Ward L, Paul A, et al. ElemNet: deep learning the chemistry of materials from only elemental composition. Sci Rep 2018;8:17593.
26. Leung K, Qi Y, Zavadil KR, et al. Using atomic layer deposition to hinder solvent decomposition in lithium ion batteries: first-principles modeling and experimental studies. J Am Chem Soc 2011;133:14741-54.
27. Yang S, Hu S, Zhao J, et al. Prediction on discharging properties of nickel–manganese materials for high-performance sodium-ion batteries via machine learning methods. Energy Technol 2022;10:2200733.
28. Chen W, Schmidt JN, Yan D, Vohra YK, Chen C. Machine learning and evolutionary prediction of superhard B-C-N compounds. npj Comput Mater 2021;7:585.
29. Guo J, Che Y, Pedersen K, Stroe D. Battery impedance spectrum prediction from partial charging voltage curve by machine learning. J Energy Chem 2023;79:211-21.
30. Bartók AP, Kondor R, Csányi G. On representing chemical environments. Phys Rev B 2013;87:184115.
31. Huo H, Rupp M. Unified representation of molecules and crystals for machine learning. Mach Learn Sci Technol 2022;3:045017.
32. Novikov IS, Gubaev K, Podryabinkin EV, Shapeev AV. The MLIP package: moment tensor potentials with MPI and active learning. Mach Learn Sci Technol 2021;2:025002.
33. Shapeev AV. Moment tensor potentials: a class of systematically improvable interatomic potentials. Multiscale Model Sim 2016;14:1153-73.
34. Drautz R. Atomic cluster expansion for accurate and transferable interatomic potentials. Phys Rev B 2019;99:014104.
35. Bai X, Zhao X, Zhang Y, et al. Dynamic stability of copper single-atom catalysts under working conditions. J Am Chem Soc 2022;144:17140-8.
36. Chen C, Ong SP. A universal graph deep learning interatomic potential for the periodic table. Nat Comput Sci 2022;2:718-28.
37. Chen C, Ye W, Zuo Y, Zheng C, Ong SP. Graph networks as a universal machine learning framework for molecules and crystals. Chem Mater 2019;31:3564-72.
38. Cheng J, Zhang C, Dong L. A geometric-information-enhanced crystal graph network for predicting properties of materials. Commun Mater 2021;2:194.
39. Gong S, Yan K, Xie T, et al. Examining graph neural networks for crystal structures: Limitations and opportunities for capturing periodicity. Sci Adv 2023;9:eadi3245.
40. Merchant A, Batzner S, Schoenholz SS, Aykol M, Cheon G, Cubuk ED. Scaling deep learning for materials discovery. Nature 2023;624:80-5.
41. Du H, Hui J, Zhang L, Wang H. Rational design of deep learning networks based on a fusion strategy for improved material property predictions. J Chem Theory Comput 2024;20:6756-71.
42. Cheng D, Sha W, Han Q, et al. ACGNet: an interpretable attention crystal graph neural network for accurate oxidation potential prediction. Electrochim Acta 2024;473:143459.
43. Xie T, Grossman JC. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys Rev Lett 2018;120:145301.
44. Louis SY, Zhao Y, Nasiri A, et al. Graph convolutional neural networks with global attention for improved materials property prediction. Phys Chem Chem Phys 2020;22:18141-8.
45. Satorras VG, Hoogeboom E, Welling M. E(n) equivariant graph neural networks. arXiv. [Preprint.] Feb 16, 2022 [accessed on 2024 Oct 21]. Available from: https://doi.org/10.48550/arXiv.2102.09844.
46. Zhang D, Bi H, Dai FZ, Jiang W, Zhang L, Wang H. DPA-1: pretraining of attention-based deep potential model for molecular simulation. arXiv. [Preprint.] Sep 15, 2023 [accessed on 2024 Oct 21]. Available from: https://doi.org/10.48550/arXiv.2208.08236.
47. Zhang D, Liu X, Zhang X, et al. DPA-2: towards a universal large atomic model for molecular and material simulation. arXiv. [Preprint.] Dec 24, 2023 [accessed on 2024 Oct 21]. Available from: https://arxiv.org/html/2312.15492v1.
48. Chen C, Zuo Y, Ye W, Li X, Ong SP. Learning properties of ordered and disordered materials from multi-fidelity data. Nat Comput Sci 2021;1:46-53.
49. Chen C, Ong SP. AtomSets as a hierarchical transfer learning framework for small and large materials datasets. npj Comput Mater 2021;7:639.
50. Li Y, Wu Y, Han Y, et al. Local environment interaction-based machine learning framework for predicting molecular adsorption energy. J Mater Inf 2024;4:4.
51. Zhao X, Liu Y. Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction. J Am Chem Soc 2021:9423-8.
52. Cao Z, Magar R, Wang Y, Barati Farimani A. MOFormer: self-supervised transformer model for metal-organic framework property prediction. J Am Chem Soc 2023;145:2958-67.
53. Jain A, Ong SP, Hautier G, et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater 2013;1:011002.
54. The Materials Project. Available from: https://www.Materialsproject.Org/. [Last accessed on 21 Oct 2024].
55. Kirklin S, Saal JE, Meredig B, et al. The open quantum materials database (OQMD): assessing the accuracy of DFT formation energies. npj Comput Mater 2015;1:BFnpjcompumats201510.
56. Kim M, Kang S, Gyu Park H, Park K, Min K. Maximizing the energy density and stability of Ni-rich layered cathode materials with multivalent dopants via machine learning. Chem Eng J 2023;452:139254.
57. Lundberg S, Lee SI. A unified approach to interpreting model predictions. arXiv. [Preprint.] Nov 25, 2017 [accessed on 2024 Oct 22]. Available from: https://doi.org/10.48550/arXiv.1705.07874.
58. Chen H, Covert IC, Lundberg SM, Lee S. Algorithms to estimate Shapley value feature attributions. Nat Mach Intell 2023;5:590-601.
59. Hu Y, Zhang Y, Wen B, Dai F. Grain boundary engineering in Nickel-rich cathode: a combination of high-throughput first-principles and interpretable machine learning study. Acta Mater 2024;276:120144.
60. Jia Y, Zhang R, Fang C, Zheng J. Interpretable machine learning to accelerate the analysis of doping effect on Li/Ni exchange in Ni-rich layered oxide cathodes. J Phys Chem Lett 2024;15:1765-73.
61. Dunn A, Wang Q, Ganose A, Dopp D, Jain A. Benchmarking materials property prediction methods: the Matbench test set and Automatminer reference algorithm. npj Comput Mater 2020;6:138.
62. MatBench. Available from: https://matbench.materialsproject.org/. [Last accessed on 22 Oct 2024].
63. Behler J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J Chem Phys 2011;134:074106.
64. Valle M, Oganov AR. Crystal fingerprint space - a novel paradigm for studying crystal-structure sets. Acta Crystallogr A 2010;66:507-17.
65. Kim S, Noh J, Jin T, Lee J, Jung Y. A structure translation model for crystal compounds. npj Comput Mater 2023;9:1094.
66. Isayev O, Oses C, Toher C, Gossett E, Curtarolo S, Tropsha A. Universal fragment descriptors for predicting properties of inorganic crystals. Nat Commun 2017;8:15679.
67. Dinic F, Voznyy O. Unconstrained machine learning screening for new Li-ion cathode materials enhanced by class balancing. Adv Theory Simul 2023;6:2300081.
68. Gilmer J, Schoenholz SS, Riley PF, Vinyals O, Dahl GE. Neural message passing for Quantum chemistry. In. Proceedings of the 34th International Conference on Machine Learning; Sydney, Australia. 2017. pp. 1263-72. Available from: https://proceedings.mlr.press/v70/gilmer17a. [Last accessed on 22 Oct 2024]
69. Jørgensen PB, Jacobsen KW, Schmidt MN. Neural message passing with edge updates for predicting properties of molecules and materials. arXiv. [Preprint.] Jun 8, 2018 [accessed on 2024 Oct 22]. Available from: https://doi.org/10.48550/arXiv.1806.03146.
70. He X, Wu J, Zhu Z, et al. Chemical and structural evolutions of Li–Mn-rich layered electrodes at different current densities. Energy Environ Sci 2022;15:4137-47.
71. Choudhary K, Decost B. Atomistic line graph neural network for improved materials property predictions. npj Comput Mater 2021;7:650.
72. Wang H, Zhang L, Han J, E W. DeePMD-kit: a deep learning package for many-body potential energy representation and molecular dynamics. Comput Phys Commun 2018;228:178-84.
73. Zeng J, Zhang D, Lu D, et al. DeePMD-kit v2: a software package for deep potential models. J Chem Phys 2023;159:054801.
74. Jia W, Wang H, Chen M, Lu D, Lin L, Car R. Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning. In. SC20: International Conference for High Performance Computing, Networking, Storage and Analysis; 2020 Noc 09-19; Atlanta, USA. IEEE; 2020. pp. 1-14.
75. Deng B, Zhong P, Jun K, et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat Mach Intell 2023;5:1031-41.
76. Batatia I, Péter Kovács D, Simm GNC, Ortner C, Csányi G. MACE: higher order equivariant message passing neural networks for fast and accurate force fields. arXiv. [Preprint.] Jan 26, 2023 [accessed on 2024 Oct 22]. Available from: https://doi.org/10.48550/arXiv.2206.07697.
77. Genreith-Schriever AR, Alexiu A, Phillips GS, et al. Jahn-Teller distortions and phase transitions in LiNiO2: insights from ab initio molecular dynamics and variable-temperature X-ray diffraction. Chem Mater 2024;36:2289-303.
78. Chen L, Zhang W, Nie Z, Li S, Pan F. Generative models for inverse design of inorganic solid materials. J Mater Inf 2021;1:4.
79. Ren Z, Tian SIP, Noh J, et al. An invertible crystallographic representation for general inverse design of inorganic crystals with targeted properties. Matter 2022;5:314-35.
80. Zeni C, Pinsler R, Zügner D, et al. MatterGen: a generative model for inorganic materials design. arXiv. [Preprint.] Jan 29, 2024 [accessed on 2024 Oct 22]. Available from: https://doi.org/10.48550/arXiv.2312.03687.
81. Myung S, Maglia F, Park K, et al. Nickel-rich layered cathode materials for automotive lithium-ion batteries: achievements and perspectives. ACS Energy Lett 2017;2:196-223.
82. MacNeil DD, Lu Z, Chen Z, Dahn JR. A comparison of the electrode/electrolyte reaction at elevated temperatures for various Li-ion battery cathodes. J Power Sources 2002;108:8-14.
83. Zhang J, Cao T, Lei Y, et al. Nickel-rich layered cathode LiNi0.8Co0.1Mn0.1O2 mediated by a selective lattice doping towards high-performance lithium ion battery. J Alloys Compd 2023;957:170400.
84. Pechen LS, Makhonina EV, Medvedeva AE, et al. Effect of dopants on the functional properties of lithium-rich cathode materials for lithium-ion batteries. Russ J Inorg Chem 2021;66:777-88.
85. Shen Y, Yao X, Zhang J, et al. Sodium doping derived electromagnetic center of lithium layered oxide cathode materials with enhanced lithium storage. Nano Energy 2022;94:106900.
86. Thackeray M, David W, Bruce P, Goodenough J. Lithium insertion into manganese spinels. Mater Res Bull 1983;18:461-72.
87. Thackeray M, Johnson P, de Picciotto L, Bruce P, Goodenough J. Electrochemical extraction of lithium from LiMn2O4. Mater Res Bull 1984;19:179-87.
88. Vanaphuti P, Bong S, Ma L, Ehrlich S, Wang Y. Systematic study of different anion doping on the electrochemical performance of cobalt-free lithium–manganese-rich layered cathode. ACS Appl Energy Mater 2020;3:4852-9.
89. Wang Y, Liu J, Chen T, Lin W, Zheng J. Factors that affect volume change during electrochemical cycling in cathode materials for lithium ion batteries. Phys Chem Chem Phys 2022;24:2167-75.
90. Wang YY, Song X, Liu S, Li GR, Ye SH, Gao XP. Elucidating the effect of the dopant ionic radius on the structure and electrochemical performance of Ni-rich layered oxides for lithium-ion batteries. ACS Appl Mater Interfaces 2021;13:56233-41.
91. Yang W, Zhang H. Anion doping of LiNi0.4Co0.2Mn0.4O2 cathode material for lithium-ion batteries. Rare Metal Mat Eng 2014;43:3133-7. Available from: http://www.rmme.ac.cn/rmmeen/article/abstract/20141253. [Last accessed on 22 Oct 2024]
92. McCalla E, Abakumov AM, Saubanère M, et al. Visualization of O-O peroxo-like dimers in high-capacity layered oxides for Li-ion batteries. Science 2015;350:1516-21.
93. Padhi AK, Nanjundaswamy KS, Goodenough JB. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J Electrochem Soc 1997;144:1188-94.
94. Shin Y, Persson KA. Surface morphology and surface stability against oxygen loss of the lithium-excess Li2MnO3 cathode material as a function of lithium concentration. ACS Appl Mater Interfaces 2016;8:25595-602.
95. Wu Z, Ji S, Zheng J, et al. Prelithiation activates Li(Ni0.5Mn0.3Co0.2)O2 for high capacity and excellent cycling stability. Nano Lett 2015;15:5590-6.
96. Yoon S, Choi G, Lee J, Hwang J, Kim D. (In)Coherent-bond-networks in Ni-rich layered oxides for durable lithium-ion batteries. Chem Eng J 2023;458:141472.
97. Zhang R, Wang C, Zou P, et al. Compositionally complex doping for zero-strain zero-cobalt layered cathodes. Nature 2022;610:67-73.
98. Song Z, Wang T, Yang H, et al. Promoting high-voltage stability through local lattice distortion of halide solid electrolytes. Nat Commun 2024;15:1481.
99. Wang T, Zhang R, Wu Y, et al. Engineering a flexible and mechanically strong composite electrolyte for solid-state lithium batteries. J Energy Chem 2020;46:187-90.
100. Wang Y, Nakamura S, Ue M, Balbuena PB. Theoretical studies to understand surface chemistry on carbon anodes for lithium-ion batteries: reduction mechanisms of ethylene carbonate. J Am Chem Soc 2001;123:11708-18.
101. Wu Q, McDowell MT, Qi Y. Effect of the electric double layer (EDL) in multicomponent electrolyte reduction and solid electrolyte interphase (SEI) formation in lithium batteries. J Am Chem Soc 2023;145:2473-84.
102. Yan C, Li HR, Chen X, et al. Regulating the inner Helmholtz plane for stable solid electrolyte interphase on lithium metal anodes. J Am Chem Soc 2019;141:9422-9.
103. Duan J, Huang L, Wang T, et al. Shaping the contact between Li metal anode and solid-state electrolytes. Adv Funct Mater 2020;30:1908701.
104. Tobishima S, Morimoto H, Aoki M, et al. Glyme-based nonaqueous electrolytes for rechargeable lithium cells. Electrochim Acta 2004;49:979-87.
105. Ye Y, Hu Z, Liu J, et al. Research progress of theoretical studies on polarons in cathode materials of lithium-ion batteries. Acta Phys Chim Sin 2021;37:75-87.
106. Yin YC, Yang JT, Luo JD, et al. A LaCl3-based lithium superionic conductor compatible with lithium metal. Nature 2023;616:77-83.
107. Yoshida K, Nakamura M, Kazue Y, et al. Oxidative-stability enhancement and charge transport mechanism in glyme-lithium salt equimolar complexes. J Am Chem Soc 2011;133:13121-9.
108. Rubin M, Wen SJ, Richardson T, Kerr J, von Rottkay K, Slack J. Electrochromic lithium nickel oxide by pulsed laser deposition and sputtering. Sol Energ Mat Sol C 1998;54:59-66.
109. Goodenough JB. Evolution of strategies for modern rechargeable batteries. Acc Chem Res 2013;46:1053-61.
110. Xu Z, Guo X, Song W, et al. Sulfur-assisted surface modification of lithium-rich manganese-based oxide toward high anionic redox reversibility. Adv Mater 2024;36:e2303612.
112. Zhang T, Li D, Tao Z, Chen J. Understanding electrode materials of rechargeable lithium batteries via DFT calculations. Prog Nat Sci Mater 2013;23:256-72.
113. Zheng J, Teng G, Xin C, et al. Role of superexchange interaction on tuning of Ni/Li disordering in layered Li(NixMnyCoz)O2. J Phys Chem Lett 2017;8:5537-42.
114. Zheng J, Ye Y, Liu T, et al. Ni/Li disordering in layered transition metal oxide: electrochemical impact, origin, and control. Acc Chem Res 2019;52:2201-9.
115. Lai G, Jiao J, Fang C, et al. A deep neural network interface potential for Li-Cu systems. Adv Mater Interfaces 2022;9:2201346.
116. Lai G, Zuo Y, Jiao J, et al. The mechanism of external pressure suppressing dendrites growth in Li metal batteries. J Energy Chem 2023;79:489-94.
117. Jiao J, Lai G, Zhao L, et al. Self-healing mechanism of lithium in lithium metal. Adv Sci 2022;9:e2105574.
118. Lai G, Jiao J, Fang C, et al. The mechanism of Li deposition on the Cu substrates in the anode-free Li metal batteries. Small 2023;19:e2205416.
119. Lai G, Jiao J, Yao J, Zheng J, Ouyang C. Analysis of Li metal anode by machine learning potential. J Chin Ceram Soc 2023;51:469-75.
120. Li S, Jiang M, Xie Y, Xu H, Jia J, Li J. Developing high-performance lithium metal anode in liquid electrolytes: challenges and progress. Adv Mater 2018;30:e1706375.
121. Wang T, Duan J, Zhang B, et al. A self-regulated gradient interphase for dendrite-free solid-state Li batteries. Energy Environ Sci 2022;15:1325-33.
122. Wen J, Wang T, Wang C, et al. A tailored interface design for anode-free solid-state batteries. Adv Mater 2024;36:e2307732.
123. Zunger A. Inverse design in search of materials with target functionalities. Nat Rev Chem 2018;2:BFs415700180121.
124. Yang N, Xu X, Zheng J. Independent regulation of the lithium-ion conductivity of LiF using elemental doping: a first-principles study. Phys Rev Mater 2024;8:015403.
Cite This Article

How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright














Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].