

Continuous-flow electrooxidation for scalable biomass upgrading over copper-supported CoFe Prussian blue analogues
 0
0 Abstract
Electrochemical biomass upgrading is a promising substitute for oxygen evolution reaction (OER) to generate valuable chemicals in conjunction with hydrogen generation. Pursuing highly efficient and durable electrocatalysts for significant concentration levels (≥ 50 mM) of biomass electrooxidation remains an enduring challenge. Herein, we introduce a robust Cu-supported CoFe Prussian blue analogue (CoFe PBA/CF) electrocatalyst, adept at facilitating high-concentration (50 mM) 5-hydroxymethylfurfural (HMF) oxidation into 2,5-furandicarboxylic acid (FDCA), achieving an exceptional HMF conversion (100%) with a notable FDCA yield of 98.4%. The influence of copper substrate and adsorption energy are therefore discussed. Impressively, the CoFe PBA/CF electrode sustains considerable durability in a continuous-flow electrochemical reactor designed for consecutive FDCA production, showcasing FDCA yields of 100/94% at flow rates of 0.4/0.8 mL·min-1 over 60 h’ uninterrupted electrolysis. This work provides a promising strategy to develop highly efficient and robust electrocatalysts for the consecutive production of high-value products coupled with green H2 production.

Keywords
INTRODUCTION
Hydrogen production through electrolytic water splitting is a crucial approach to achieving green and sustainable development[1-4]. However, the sluggish anodic oxygen evolution reaction (OER) kinetics and elevated overpotential pose challenges for water electrolysis, resulting in reduced hydrogen production efficiency and increased energy consumption[5-7]. Considering the significant equilibrium potential [1.23 V vs. the reversible hydrogen electrode (RHE)] and elevated overpotential (≥ 190 mV) of OER[8,9], it is necessary to substitute such anodic OER with the electrocatalysis of oxidation reactions that involve small organic molecules, which exhibit more favorable thermodynamics and economic value[10-12]. One such typical reaction is the 5-hydroxymethylfurfural (HMF) electrocatalytic oxidation reaction (HMFOR)[13-15]. The oxidation product (FDCA) serves mainly as a monomer substitute for fossil fuel-derived terephthalic acid in the production of polyethylene terephthalate (PET)[16], and is listed among the top ten most important bio-platform chemicals by the US Department of Energy[17,18].
Recently, transition metal-based electrocatalysts have received considerable attention due to the high conversion, selectivity, and stability in HMFOR[13,15,19-21]. In many studies, small volumes of dilute HMF (≤
Prussian blue analogue (PBA) derived materials have the advantages of high surface area, uniform porous structure, and structural tunability[29-31]. Studies indicate that PBA materials can be readily reconstituted into ultrathin two-dimensional (2D) hydroxyl oxide nanosheets during electrocatalysis, which is considered as the active phase for HMFOR[32]. Notably, the activity of hydroxyl oxide generated through this approach significantly surpasses that of bulk hydroxyl oxide materials, providing a substantial catalytic advantage[33-35]. Furthermore, recent studies propose that Cu can suppress the deprotonation of OH* to O* species and enhance the adsorption and oxidation of aldehyde in HMF, thereby passivating water oxidation activity and enhancing the overall efficiency of HMFOR[36-38]. Therefore, constructing a self-supported PBA catalyst grown on Cu foams is expected to yield elaborately designed catalysts with well-organized structures and highly exposed active sites, which are anticipated to accelerate the HMF adsorption and conversion into desired products[39].
In this study, we developed a self-supported CoFe PBA nanocube propagating on Cu foam (CoFe PBA/CF) via a one-step hydrothermal method for the oxidation of HMF to FDCA under high-concentration conditions (50-100 mM). The integration of Cu substrate not only perpetuates PBA structural regularity and escalates the number of surface-active sites, but also modulates the surface electronic state of Co within the catalyst. This modification augments the adsorption efficiency of HMF on the catalyst surface, thus favoring the electrooxidation process of HMF to FDCA. Moreover, a continuous flow reactor was designed to circumvent the impediments posed by diffusion limitation of high-concentration electrolyte during HMFOR and realize the consecutive production, thus fostering the practical application of the electrocatalytic conversion process from HMF to FDCA.
EXPERIMENTAL
Materials
Cobalt acetate tetrahydrate (C4H6CoO4·4H2O), polyvinylpyrrolidone, and potassium hydroxide (KOH) were purchased from Aladdin Company. Sodium citrate dihydrate (C6H5Na3O7·2H2O) and potassium ferricyanide (K3FeC6N6) were obtained from Macklin Company. Cu foam was purchased from Kunshan Guangjiayuan new material Co., Ltd. Materials 5-hydroxymethyl furfuraldehyde (HMF), 2,5-diformyl furan (DFF), 5-hydroxymethyl-2-furan carboxylic acid (HMFCA), 2,5-furandicarboxylic acid (FDCA) and
Preparation of CoFe PBA/CF
The synthesis process proceeded as follows: Initially, solution A was prepared by dissolving 0.1 g of cobalt (II) acetate tetrahydrate, 0.14 g of trisodium citrate dihydrate, and 1 g of polyvinylpyrrolidone in 6.5 mL of deionized water. Concurrently, solution B was prepared by dissolving 0.0667 g of potassium hexacyanoferrate (III) in 10 mL of deionized water. Subsequently, solution B was introduced into solution A while subject to magnetic stirring, and the resulting mixture was stirred continuously for 75 s. Following this, the solution obtained, along with a piece of cleaned Cu foam measuring 1 × 2 cm2, was transferred into a 100 mL Teflon-lined stainless-steel autoclave. The autoclave was sealed and subjected to heating at 100 °C for 16 h. Upon completion of the heating process, the autoclave was allowed to cool to room temperature. The resulting CoFe PBA supported on Cu foam was then washed successively with deionized water and ethanol, and finally dried at 60 °C.
Preparation of CoFe PBA-CF
The remaining solid from the synthesis of CoFe-PBA/CF was collected through centrifugation and subsequently dried under vacuum conditions at 60 °C overnight to obtain CoFe PBA. To prepare the catalyst dispersion, 1.5 mg of the catalysts were combined with 1,000 µL of isopropanol and 20 µL of Nafion solution. This mixture was then subjected to ultrasonication for a duration of 1.5 h. For the fabrication of the working electrode, the as-prepared catalyst dispersion was applied to half of a piece of cleaned Cu foam (1 × 2 cm2) and left to dry at room temperature to obtain CoFe PBA-CF.
Physical methods
Scanning electron microscopy (SEM) was conducted using a Hitachi S-4800. Transmission electron microscopy (TEM) characterization and elemental mapping measurements were performed on a FEI Tecnai G2 F20. X-ray powder diffraction (XRD) characterization was recorded on a Bruker Corporation D8 ADVANCE with a Cu Kα source (1.54056 Å). X-ray photoelectron spectroscopy (XPS) was conducted using a Thermo Fisher Scientific ESCALAB 25. Fourier transform infrared spectroscopy (FTIR) characterization was recorded on a Thermo Fisher Scientific Nicolet iS50.
In-situ Raman spectroelectrochemical measurement
Raman spectra were obtained utilizing a confocal Raman microscope (Horiba LabRAM HR Evolution) employing a 633 nm excitation wavelength and a 50× objective. Each displayed Raman spectrum was gathered over a 30-second collection period and represents the average of three measurements.
In-situ electrochemical impedance spectroscopy tests
In situ electrochemical impedance spectroscopy (EIS) measurements were carried out in a three-electrode system using the Corrtest electrochemical workstation (CS310X), under the same conditions as OER and HMF oxidation experiments, but without stirring. The frequency range was from 104 to 10-2 Hz, and the applied potential ranged from 1.2 V to 1.7 V vs. RHE with a 0.05 V interval. EIS measurements for OER were performed in 1 M KOH, while for HMF oxidation, they were conducted in 1 M KOH and 50 mM HMF, respectively.
High-performance liquid chromatography analysis of oxidation products
A 10 μL aliquot was periodically extracted from the electrolyte solution during chronoamperometry at
The conversion (%) of HMF, the yield (%) of FDCA and Faradaic efficiency (FE) for FDCA production were calculated based on
Where mcon represents mol of HMF consumed, mini indicates mol of HMF initial, mfor stands for mol of FDCA formed, C points to total charge passed, F denotes Faraday’s constant (96,500 C·mol-1), and n is the electron transfer number during the electrooxidation process.
Design of flow reactor
The components of the flow reactor were designed and purchased from Shanghai Chuxi Industrial Co., Ltd. The length, width, height and the wall thickness of the reactor are 4, 4, 4, and 1.5 cm, respectively.
Electrocatalytic experiments
Electrochemical measurements (OER and HMFOR) were conducted using a Corrtest electrochemical workstation (CS310X) potentiostat with a three-electrode configuration in 1 M KOH solution. The Hg/HgO electrode served as the reference electrode. All potentials mentioned in this paper were referenced to the RHE through calibration. The electrochemical experiments (OER and HMFOR) were performed in a 10 mL 1.0 M KOH aqueous solution with and without 50 mM HMF. The cyclic voltammetry (CV) tests are conducted on the CoFe PBA/CF and CoFe PBA-CF catalysts in an alkaline electrolyte to induce their reconstruction. Under alkaline conditions and electric drive etching, the N=C–Fe groups gradually break and leach out. When the CV curves overlap completely (after ~100 cycles), the material reconstruction is complete. Linear sweep voltammetry (LSV) was conducted using the three-electrode configuration at a scan rate of 2 mV·s-1.
For the two-electrode electrolysis in the electrochemical continuous flow reactor, CoFe PBA/CF was used as the anode catalyst, and platinum mesh was used as the cathode catalyst at 1.4 V. The double-layer capacitance (Cdl) was determined by analyzing the CV curves obtained in the non-faradaic region at different scan rates.
Density functional theory
All density functional theory (DFT) calculations were conducted by using the Vienna Ab Initio Simulation Package (VASP)[40,41], using the generalized gradient approximation (GGA) of Perdew-Burke-Ernzerhof (PBE) for the exchange-correction functional[42]. The effective U for Co (Ueff = 3.7 eV) was obtained by analyzing the highly localized 3d orbitals of metal ions[43,44]. A cut-off energy of 400 eV was employed. The energy change was < 10-5 eV per atom and the iterative process considered was converged when the force on the atom was < 0.03 eV·Å-1. Spin-polarized calculations were incorporated to describe the magnetic properties of Co element.
The CoOOH (001) surface was modeled with p (2 × 2) unit cell and the Brillouin-zone integrations were performed using a (3 × 3 × 1) Monkhorst-Pack mesh during the optimization. The optimized (CoOOH) 4 cluster was placed onto the Cu(111) with p (5 × 5) unit cell of three layers, obtaining the CoOOH/Cu(111) model. The model was simulated using extensive ab initio molecular dynamics (AIMD) simulation, and the oscillation amplitude of total energy gets small, indicating that the steady state has been reached and the great stability was confirmed. One OH group was removed to establish surfaces with oxygen vacancy [CoOOH-Ov and CoOOH-Ov/Cu(111)].
The Gibbs free energies (G) at 298.15 K and 1 atm were calculated by:
The EDFT and EZPE represent the total energy obtained from DFT optimization and the zero-point vibrational energy using the harmonic approximation, respectively[45]. CV, T and S are the heat capacity, kelvin temperature and the entropy, respectively.
RESULTS AND DISCUSSION
Structural and morphological characterizations
The CoFe PBA/CF was synthesized through a hydrothermal synthesis method, exhibiting an evenly distributed and meticulously arrayed continuum of the nanocubes with a diameter of approximately 500 nm [Figure 1A and B]. A high-resolution transmission electron microscope (HRTEM) image reveals these structures uphold a defective cube orientation [Figure 1C] and a lattice spacing of 5.12 Å is observed, which can be attributed to the (200) crystal planes inherently presenting in CoFe PBA (Figure 1C, inset). Energy-dispersive X-ray spectroscopy (EDX) mapping [Figure 1D] illustrates the even distribution of elements on the nanocube, and the inclusion of Cu elements confirms that Cu foam serves not only a supporting role but also facilitates the overall electronic environment of the catalyst. For comparative analysis, powdered CoFe PBA was synthesized using the same method but without growth on Cu foam [Supplementary Figures 1 and 2]. Drip-casting CoFe PBA powder onto the surface of Cu foam (CoFe PBA-CF) resulted in uneven distribution and weak binding between the PBA nanocubes and Cu foam. This comparison suggests that the incorporation of a Cu substrate significantly regularizes the arrangement of PBA nanocubes.
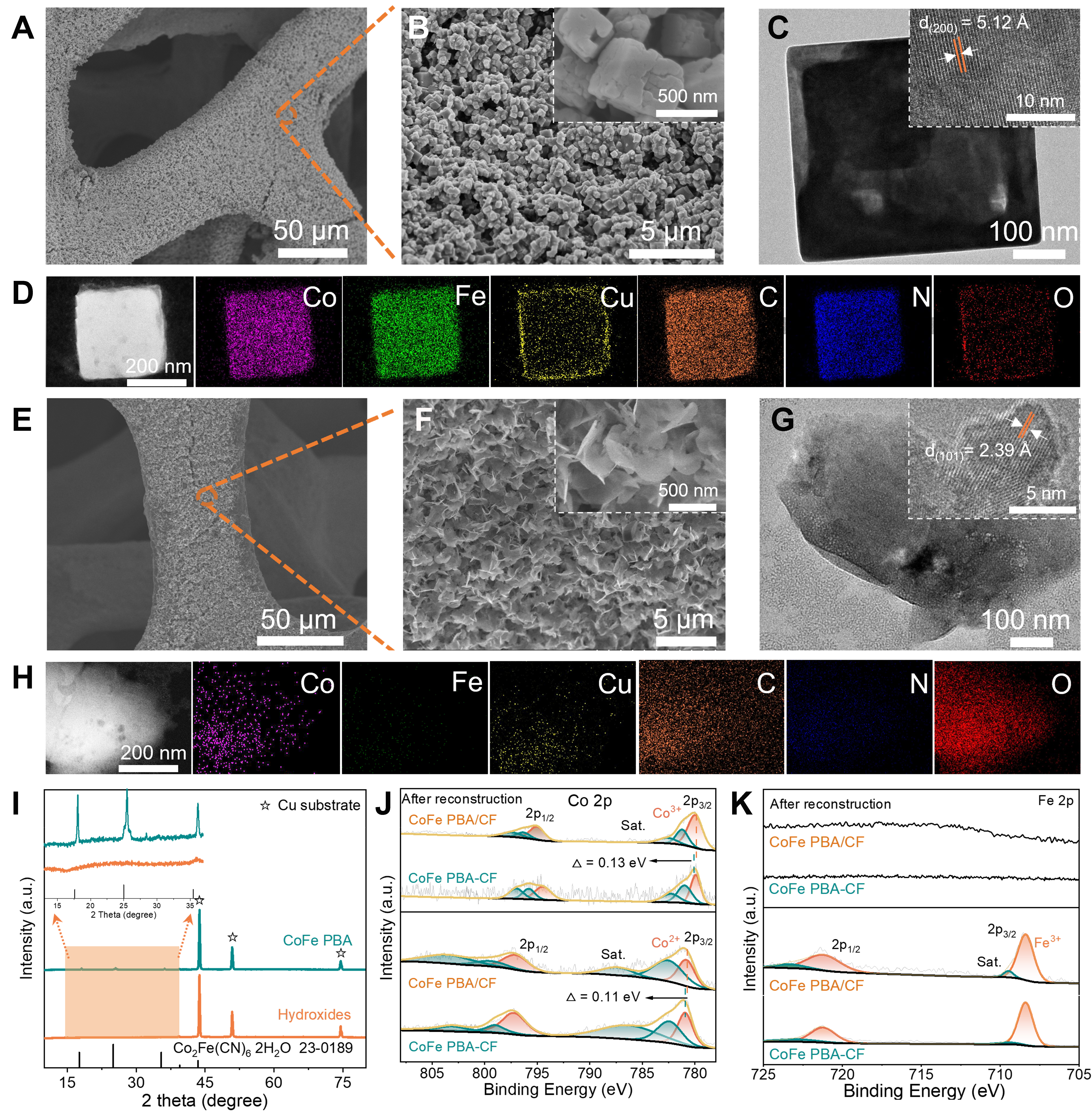
Figure 1. Material characterizations of the CoFe PBA/CF sample. (A and B) SEM, (C) HRTEM images, and (D) EDX mapping; (E and F) SEM, (G) HRTEM images, and (H) EDX mapping after reconstruction in KOH solution; (I) XRD pattern and (J and K) XPS spectra of Co 2p and Fe 2p for CoFe PBA/CF and CoFe PBA-CF samples before and after reconstruction in KOH solution. CoFe PBA/CF: Cu-supported CoFe Prussian blue analogue; SEM: scanning electron microscopy; HRTEM: high-resolution transmission electron microscope; EDX: energy-dispersive X-ray spectroscopy; XRD: X-ray powder diffraction; XPS: X-ray photoelectron spectroscopy.
It is well-established that the catalytic efficacy of transition metal derivatives becomes active only after undergoing reconstruction to transform into hydroxides[46,47]. We conducted a detailed comparison of the structural evolution between the CoFe PBA/CF and CoFe PBA-CF electrodes after electric-driven reconstruction in KOH solution [Figure 1E-G, Supplementary Figure 3]. The initially well-aligned PBA nanocubes experienced complete reconstruction, leading to the formation of distinct Co(OH)2 nanosheets. Element mapping [Figure 1H] indicates the presence of Co and Cu elements, while the levels of Fe and N elements significantly decrease in the reconstructed nanosheets. Accompanied by XRD [Figure 1I], FTIR
HMFOR performance of the surface reconstructed CoFe PBA/CF electrode
An H-type electrochemical cell, utilizing 1.0 M KOH as the cathodic electrolyte and a solution containing 50 mM HMF in 1.0 M KOH as the anodic electrolyte, was employed to assess the electrochemical HMFOR performance of CoFe PBA/CF and CoFe PBA-CF [Supplementary Figure 7]. LSV curves were measured at a scan rate of 2 mV·s-1 to compare the competing reactions between OER and HMFOR [Figure 2A, Supplementary Figure 8]. The self-supported CoFe PBA/CF electrode exhibited the lowest potential of 1.20/1.38 V vs. RHE at 10/100 mA·cm-2 for HMF oxidation (50 mM in 1 M KOH) compared to the CoFe PBA-CF electrode (1.30/1.53 V vs. RHE) and the condition without HMF (1.45/1.60 V vs. RHE). To further elucidate the interfacial electrochemical behavior during the catalytic process, in situ EIS was conducted, and the corresponding Bode phase plots are presented in Figure 2B and C. The low-frequency region (10-2 to 101 Hz) is related to the nonhomogeneous charge distribution, specifically the appearance of oxidation species at the electrode interface[50,51]. A peak is found in the low-frequency region at 1.45 V either with or without HMF, indicating OER occurs at the electrode surface. The introduction of HMF results in an additional peak at 1.25 V, indicating HMF oxidation occurs at this voltage level. Combined LSV and EIS analyses confirm that the suitable electrocatalytic HMFOR range is 1.20 to 1.45 V, during which the occurrence of OER side reactions is avoided.
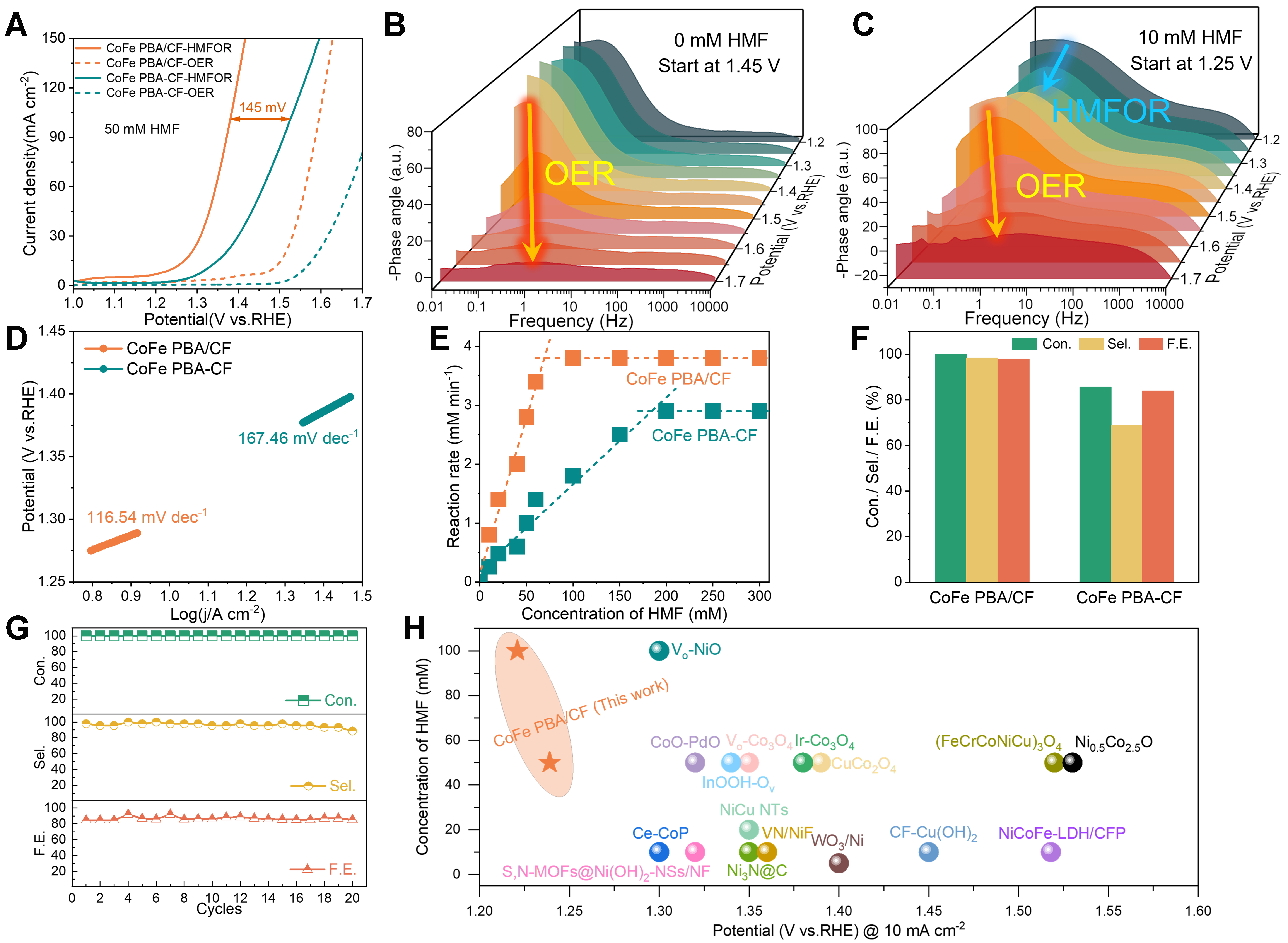
Figure 2. (A) LSV curves of CoFe PBA/CF and CoFe PBA-CF samples in 1M KOH with 50 mM HMF and without HMF; (B and C) Bode phase plots of CoFe PBA/CF sample; (D) Tafel plots of CoFe PBA/CF and CoFe PBA-CF electrodes; (E) HMF reaction rate of CoFe PBA/CF and CoFe PBA-CF electrodes with varying HMF concentrations; (F) Comparison of HMF conversion, FDCA yield, and FE between CoFe PBA/CF and CoFe PBA-CF; (G) Durability test for CoFe PBA/CF sample; (H) Comparison of overpotential for CoFe PBA/CF with other reported catalysts. LSV: Linear sweep voltammetry; CoFe PBA/CF: Cu-supported CoFe Prussian blue analogue; HMF: 5-hydroxymethylfurfural; FDCA: 2,5-furandicarboxylic acid; FE: Faradaic efficiency.
Moreover, the CoFe PBA/CF electrode also delivered a considerably smaller Tafel slope at 116.54 mV·dec-1 compared to CoFe PBA-CF at 167.46 mV·dec-1, suggesting that the intrinsic HMFOR reaction kinetics could be enhanced via the chemical interaction between the PBA nanocubes and the Cu foam substrate [Figure 2D]. The reaction rates of HMF with various concentrations were further evaluated to ascertain the intrinsic activity of CoFe PBA towards HMFOR [Figure 2E, Supplementary Figure 9]. For CoFe PBA/CF, a steady conversion rate of 3.8 mM·min-1 was achieved when the HMF concentration was approximately
HPLC was utilized to analyze and quantify the oxidation products following HMFOR. At the potential of 1.45 V, HMF underwent rapid conversion to FDCA, ultimately attaining 100% HMF conversion with a yield of 98.4% and 98% FE towards FDCA production [Figure 2F]. By contrast, the powder CoFe PBA-CF can only achieve an 85.7% conversion, 69% selectivity, and 84% FE. The durability of CoFe PBA/CF was further clarified by conducting cycling experiments [Figure 2G, Supplementary Figure 11]. Impressively, even after 20 cycles, HMF conversion remained stable at 100%, and FDCA productivity sustained at approximately 90%. This stability suggests that CoFe PBA/CF demonstrates high activity, excellent FDCA selectivity, and remarkable catalytic recyclability owing to its regular structure and complete exposure of active sites, even in a strong alkali environment. Additionally, we monitored the hydrogen evolution during the HMFOR process [Supplementary Video 1], observing a significant hydrogen generation. The calculated hydrogen generation rate is approximately 1.1 mL·min-1, with the transferred charge quantity closely matching the detected anode values. Notably, the superior electrocatalytic activity observed for CoFe PBA/CF surpasses those previously reported transition oxides derivate catalysts for HMFOR to FDCA in the last three years [Supplementary Table 2, Figure 2H].
The reaction mechanism and DFT calculations
Two possible reaction pathways for HMF oxidation are illustrated in Figure 3A, including the initial alcohol hydroxyl oxidation to DFF and the aldehyde oxidation to HMFCA, respectively[52,53]. As the reaction proceeded, the content of HMF diminished gradually, while the content of intermediates, namely HMFCA and FFCA, increased at initial stage and declined towards the end of the reaction [Figure 3B, Supplementary Figure 12]. This outcome suggests that the reaction follows an HMF-HMFCA-FFCA-FDCA pathway with the CoFe PBA/CF catalyst. In addition, we conducted a quantitative analysis of the oxidative products. Chronoamperometric electrolysis was performed at a constant voltage of 1.45 V to input a theoretical charge of 289.5 C with an HMF concentration of 50 mM [Figure 3C]. It is observed that the content of HMF considerably drops during the initial 100 C, accompanied by the formation of HMFCA and FDCA. Notably, the intermediate FFCA maintained a very low presence throughout the reaction, hinting that the transition from HMFCA to FDCA occurred rapidly.
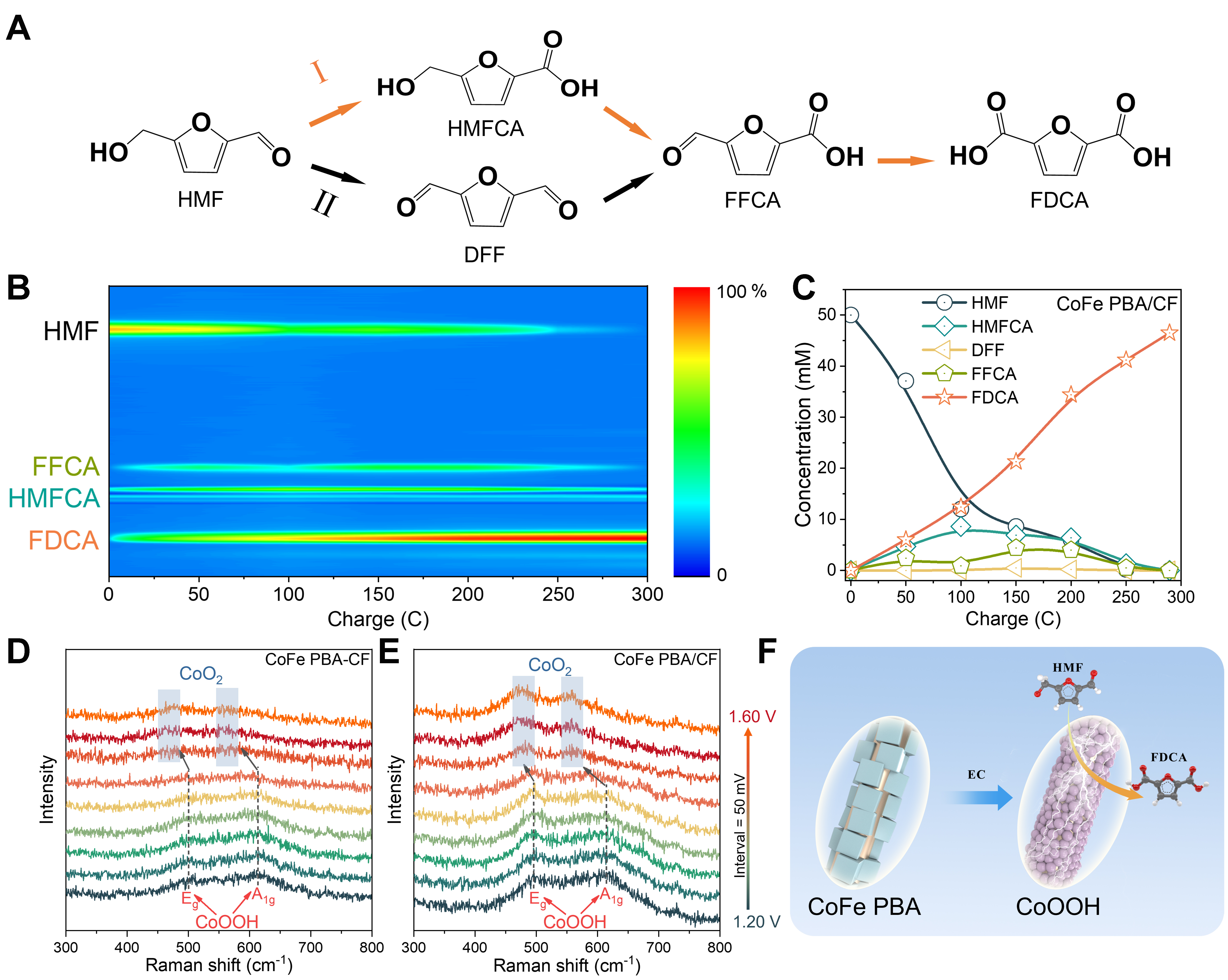
Figure 3. (A) Two possible reaction pathways for HMF electrooxidation; (B and C) Concentration changes of the reactant and products during HMFOR at 1.45 V; (D and E) In situ Raman spectra of HMFOR process on CoFe PBA-CF and CoFe PBA/CF sample; (F) Illustration of transformation of electrocatalyst and the electrooxidation of HMF on reactive sites. HMF: 5-hydroxymethylfurfural; HMFOR: 5-hydroxymethylfurfural electrocatalytic oxidation reaction; CoFe PBA/CF: Cu-supported CoFe Prussian blue analogue.
In-situ electrochemical Raman spectroscopy was used to identify the active phase of catalysts during the HMFOR[32,54]. The experiments involved a preliminary 10-minute electrolysis before measuring Raman spectra to ensure the conditions as close as possible to the true reaction environment. Two broad spectral features at 497 and 609 cm-1 were detected on both CoFe PBA-CF and CoFe PBA/CF samples, which can be attributed to the Eg and A1g vibration modes of Co3+–O bonds, respectively [Figure 3D and E]. The higher relative intensity observed on the CoFe PBA/CF electrode indicates that CoFe PBA in-situ grown on the Cu substrate tends to reconstruct more thoroughly, forming abundant active sites when driven by the potential in alkaline conditions. It is noticed that the peaks remained unchanged until the potential reached 1.45 V, suggesting that the active phase for HMFOR exclusively involves CoOOH when the current density reaches 100 mA·cm-2 (1.380 V). Moreover, these peaks gradually red-shifted to 474 and 558 cm-1 when the potential reached 1.50 V and weakened further upon incrementing potential. This red shift can be assigned to the
DFT simulations were conducted to deepen our understanding of the promotion effect of the Cu substrate on HMFOR. Observations revealed a lower Bader charge for the Co site on CoOOH/Cu(111) (+0.670 |e|) compared to that on the CoOOH surface (+1.178 |e|), implying electron transfer from the Cu substrate to the Co sites [Figure 4A and B]. Simultaneously, the region around the Cox+ site of CoOOH/Cu(111) exhibited the most positive electrostatic potential value, making it easier for reactants to adsorb. Thus, we calculated the adsorption energies of HMF at different Cox+ sites. Considering that the oxygen atoms of formyl (-CHO) in HMF act as electron acceptors, they show a preference for adsorption on electron-rich Co sites from the CoOOH/Cu(111) surface. Notably, the adsorption energy of HMF on CoOOH/Cu(111) is -1.00 eV, significantly greater than that on the CoOOH (-0.52 eV) surface [Supplementary Figures 13-15]. This observation suggests that the introduction of Cu substrate facilitates the absorption of HMF molecules.
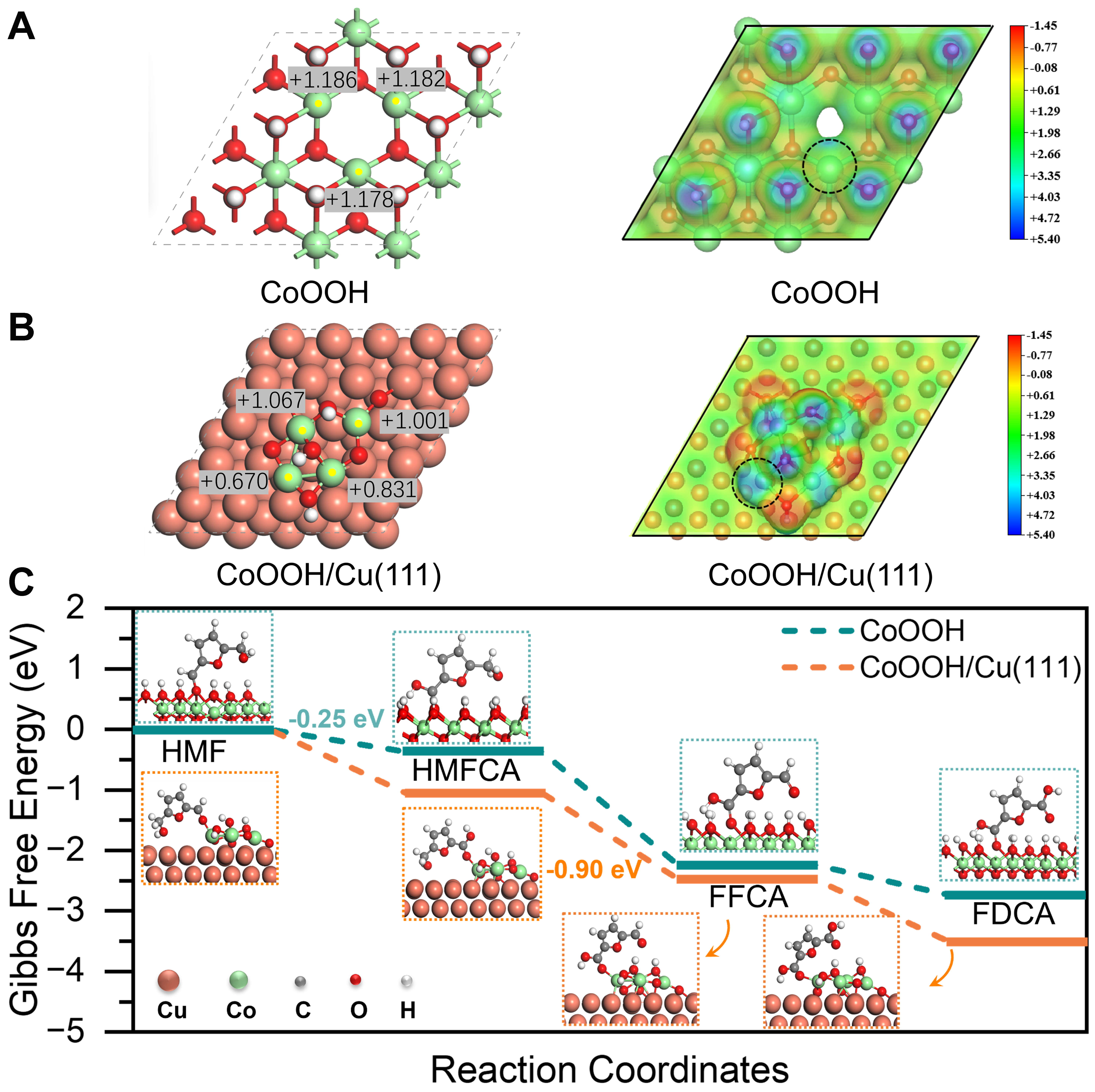
Figure 4. (A and B) Top-view diagrams of constructed models for CoOOH, CoOOH/Cu(111), including the corresponding Bader charge transfer (|e|) and electrostatic potentials with an isosurface value of 0.003 e/Bohr3; (C) Free energy diagram for HMFOR on both CoOOH and CoOOH/Cu(111), illustrating the adsorption configurations of intermediates. HMFOR: 5-hydroxymethylfurfural electrocatalytic oxidation reaction.
We further conducted studies on Gibbs free energy evolutions for HMFOR to FDCA conversion both on CoOOH and CoOOH/Cu(111) surfaces [Figure 4C] to elucidate the differences in electrocatalytic performance for HMF oxidation. The results demonstrate that the ΔG values for the HMFOR elementary processes on both CoOOH and CoOOH/Cu(111) are thermodynamically favorable. The larger free energy gap observed in the step from HMFCA to FFCA on CoOOH/Cu(111) (-0.90 eV), compared to that for HMF to HMFCA on CoOOH (-0.25 eV), suggests that the active site exhibits reduced charge transfer, catalyzes the dehydrogenation step of HMF and thereby modifies the reaction kinetics. Therefore, CoOOH/Cu(111), with its large Gibbs free energy gap for the potential path and adsorption energy of HMF, is more favorable for the conversion of HMF compared to CoOOH. Based on the theoretical results, we can infer that the electronic redistribution between Co and Cu facilitates the catalytic progress for HMFOR, which is consistent with the experimental observations.
Continuous flow electrooxidation of HMF to FDCA
Considering the industrial applications, the production of FDCA should ideally operate continuously within a practical reactor[19,21,57,58]. Importantly, improving the mass transfer of the electrolyte is critical to the electrochemical reaction, particularly when the reactant concentration is high (≥ 50 mM). In a traditional H-type cell, the reactant species on the active sites are not easy to refresh timely for fast conversion and the reaction of different benches of feedstock is intermittent, thus significantly reducing the production efficiency. To achieve consecutive and efficient production of FDCA from concentrated HMF solution, we have developed a continuous-flow electrochemical reactor (CFER) [Figure 5A]. The KOH and HMF solutions are independently delivered and subsequently mixed in a tee valve before being pumped into the CFER. This setup enables the rapid conversion of the reactive substrate into the targeted product within the CFER, effectively preventing the degradation of HMF in the KOH solution. Notably, both the conversion and selectivity of HMF to FDCA remained remarkably high at close to 100% at the flow rate of 0.4 mL·min-1 [Figure 5B]. The procedure entailing the continuous oxidation of high-concentration HMF to FDCA via the CFER was documented through videography [Supplementary Video 2]. Observations from the video, in conjunction with Supplementary Figure 16, divulge that the yellow-hued HMF solution amalgamates with KOH in the tee valve and subsequently engenders a colorless FDCA solution in CFER.
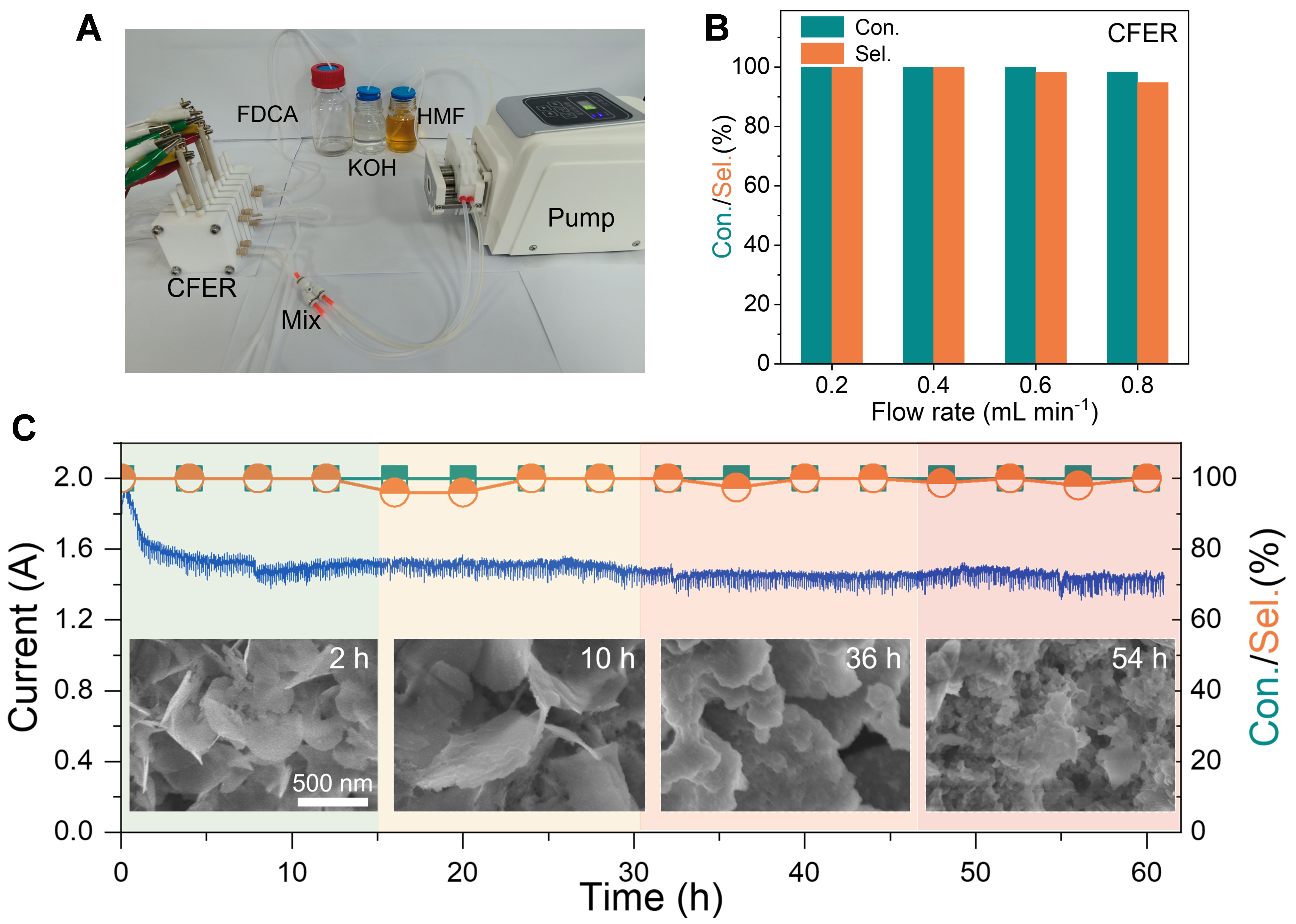
Figure 5. (A) Photographic image of the CFER for HMFOR; (B) HMF conversion and FDCA selectivity at various flow rates; (C) Durability test of CoFe-PBA/CF in the CFER at the potential of 1.4 V with 50 mM HMF in 1 M KOH solution as the electrolyte. CFER: Continuous-flow electrochemical reactor; HMFOR: 5-hydroxymethylfurfural electrocatalytic oxidation reaction; HMF: 5-hydroxymethylfurfural; FDCA: 2,5-furandicarboxylic acid; CoFe PBA/CF: Cu-supported CoFe Prussian blue analogue.
Furthermore, an escalated flow rate (0.8 mL·min-1) delivered continued HMF conversions of approximately 99%, while the selectivity for FDCA remained stable at around 94%. Such superior performance infers that the CoFe PBA/CF catalyst is capable of swiftly converting HMF, thereby demonstrating its capacity to effectively handle high concentrations of HMF during the reaction process. We also assessed the durability of the CoFe PBA/CF electrodes [Figure 5C, Supplementary Figure 17]. Even after enduring over 60 h of continuous electrolysis within the CFER, these electrodes still maintained impressive HMF conversion rates (close to 100%) and FDCA selectivity (97%), signifying the durability and promising productivity potential. Moreover, we monitored structural transformations of the PBA over various time periods during the HMFOR reaction to explore the correlation between its elevated stability and structural changes (Figure 5C, inset, Supplementary Figures 18 and 19). CoFe PBA/CF was transformed into Co(OH)2 nanosheets and demonstrated prolonged structural stability during HMFOR. Accordingly, these results suggest the immense potential of our CoFe PBA/CF electrodes and the engineered CFER for facilitating the continuous, large-scale production of FDCA under industrial conditions.
CONCLUSIONS
In summary, a robust self-supported CoFe PBA/CF electrocatalyst has been constructed via a hydrothermal process. The self-supported CoFe PBA/CF demonstrates superb HMFOR electrocatalytic performance, achieving a remarkable 98.4% yield of FDCA and a 98% FE when subjected to high-concentration HMF solutions (≥ 50 mM), representing a significant enhancement compared to current low-dose HMF conversion processes. DFT theoretical calculations and series characterizations show that the introduction of Cu substrate not only improves PBA structural regularity and the number of surface-active sites, but also regulates the surface electronic state of Co in the catalyst, thereby facilitating the adsorption and conversion of HMF. Impressively, a continuous flow electrochemical reactor is designed to accelerate the diffusion, thereby enabling operation at high reaction rates. This design achieves a yield of FDCA close to 97% during the continuous 60-hour reaction. Our contribution paves the way for biomass upgrading to produce value-added products by constructing high-performance self-supported electrodes and coupling them with a continuous flow reactor.
DECLARATIONS
Authors’ contributions
Designed, prepared and revised the manuscript: Zhang B, Hu C, Zhang H, Yuan P
Discussion and preparation of the manuscript: Zhang B, Xiao T, Hu C, Liu Z, Chen P, Zhao Z, Lai D, Huang J, Zhang H, Bao X, Yuan P
Availability of data and materials
The raw data supporting the findings of this study are available within this Article and its Supplementary Materials. Further data is available from the corresponding authors upon request.
Financial support and sponsorship
This work is supported by the National Key R&D Project (2022YFB4004100), National Natural Science Foundation of China, Pilot Group Program of the Research Fund for International Senior Scientists (22250710676), National Natural Science Foundation of China (22078064, 22304028), and Natural Science Foundation of Fujian Province (2021J02009).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
1. Gao, G.; Zhu, G.; Chen, X.; Sun, Z.; Cabot, A. Optimizing Pt-based alloy electrocatalysts for improved hydrogen evolution performance in alkaline electrolytes: a comprehensive review. ACS. Nano. 2023, 17, 20804-24.
2. Wang, J.; Yue, X.; Yang, Y.; et al. Earth-abundant transition-metal-based bifunctional catalysts for overall electrochemical water splitting: a review. J. Alloys. Compd. 2020, 819, 153346.
3. Yang, Q.; Liu, H.; Yuan, P.; et al. Single carbon vacancy traps atomic platinum for hydrogen evolution catalysis. J. Am. Chem. Soc. 2022, 144, 2171-8.
4. Liu, W.; Niu, X.; Tang, J.; et al. Energy-efficient anodic reactions for sustainable hydrogen production via water electrolysis. Chem. Synth. 2023, 3, 44.
5. Zhuang, L.; Jia, Y.; Liu, H.; et al. Sulfur-modified oxygen vacancies in iron-cobalt oxide nanosheets: enabling extremely high activity of the oxygen evolution reaction to achieve the industrial water splitting benchmark. Angew. Chem. Int. Ed. Engl. 2020, 59, 14664-70.
6. Wang, B.; Chen, X.; He, Y.; et al. Fe2O3/P-doped CoMoO4 electrocatalyst delivers efficient overall water splitting in alkaline media. Appl. Catal. B. Environ. 2024, 346, 123741.
7. Wang, C.; Jiang, X.; Wang, Y.; Tang, Y.; Zhou, J.; Fu, G. Recent advances in nonmetallic modulation of palladium-based electrocatalysts. Chem. Synth. 2023, 3, 8.
8. Zhang, J.; Zhang, Q.; Feng, X. Support and interface effects in water-splitting electrocatalysts. Adv. Mater. 2019, 31, e1808167.
9. Walter, M. G.; Warren, E. L.; McKone, J. R.; et al. Solar water splitting cells. Chem. Rev. 2010, 110, 6446-73.
10. Urbańczyk, E.; Maciej, A.; Stolarczyk, A.; Basiaga, M.; Simka, W. The electrocatalytic oxidation of urea on nickel-graphene and nickel-graphene oxide composite electrodes. Electrochim. Acta. 2019, 305, 256-63.
11. Hu, S.; Wang, B.; Ma, Y.; Li, M.; Zhang, L.; Huang, Z. Ultrathin bismuth tungstate nanosheets as an effective photo-assisted support for electrocatalytic methanol oxidation. J. Colloid. Interface. Sci. 2019, 552, 179-85.
12. Barwe, S.; Weidner, J.; Cychy, S.; et al. Electrocatalytic oxidation of 5-(hydroxymethyl)furfural using high-surface-area nickel boride. Angew. Chem. Int. Ed. Engl. 2018, 57, 11460-4.
13. Zhang, N.; Zou, Y.; Tao, L.; et al. Electrochemical oxidation of 5-hydroxymethylfurfural on nickel nitride/carbon nanosheets: reaction pathway determined by in situ sum frequency generation vibrational spectroscopy. Angew. Chem. Int. Ed. Engl. 2019, 58, 15895-903.
14. Xu, H.; Xin, G.; Hu, W.; et al. Single-atoms Ru/NiFe layered double hydroxide electrocatalyst: efficient for oxidation of selective oxidation of 5-hydroxymethylfurfural and oxygen evolution reaction. Appl. Catal. B. Environ. 2023, 339, 123157.
15. Luo, R.; Li, Y.; Xing, L.; et al. A dynamic Ni(OH)2-NiOOH/NiFeP heterojunction enabling high-performance E-upgrading of hydroxymethylfurfural. Appl. Catal. B. Environ. 2022, 311, 121357.
16. Zhang, B.; Fu, H.; Mu, T. Hierarchical NiSx/Ni2P nanotube arrays with abundant interfaces for efficient electrocatalytic oxidation of 5-hydroxymethylfurfural. Green. Chem. 2022, 24, 877-84.
17. Bozell, J. J.; Petersen, G. R. Technology development for the production of biobased products from biorefinery carbohydrates - the US Department of Energy’s “Top 10” revisited. Green. Chem. 2010, 12, 539-54.
18. Werpy, T.; Petersen, G.; Aden, A.; et al. Top value added chemicals from biomass. Volume 1: results of screening for potential candidates from sugars and synthesis gas. 2004. Available from: https://www.rivertop.com/files/bioenergy/pdfs/35523.pdf. [Last accessed on 29 Aug 2024]
19. Wang, H.; Zhou, Y.; Tao, S. CoP-CoOOH heterojunction with modulating interfacial electronic structure: a robust biomass-upgrading electrocatalyst. Appl. Catal. B. Environ. 2022, 315, 121588.
20. Zhou, P.; Lv, X.; Tao, S.; et al. Heterogeneous-interface-enhanced adsorption of organic and hydroxyl for biomass electrooxidation. Adv. Mater. 2022, 34, e2204089.
21. Wang, H.; Zhang, J.; Tao, S. Nickel oxide nanoparticles with oxygen vacancies for boosting biomass-upgrading. Chem. Eng. J. 2022, 444, 136693.
22. Nie, J.; Liu, H. Efficient aerobic oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran on manganese oxide catalysts. J. Catal. 2014, 316, 57-66.
23. Nie, J.; Xie, J.; Liu, H. Activated carbon-supported ruthenium as an efficient catalyst for selective aerobic oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran. Chinese. J. Catal. 2013, 34, 871-5.
24. Wei, Y.; Zhang, Y.; Chen, Y.; et al. Crystal faces-tailored oxygen vacancy in Au/CeO2 catalysts for efficient oxidation of HMF to FDCA. ChemSusChem 2022, 15, e202101983.
25. Vuyyuru, K. R.; Strasser, P. Oxidation of biomass derived 5-hydroxymethylfurfural using heterogeneous and electrochemical catalysis. Catal. Today. 2012, 195, 144-54.
26. Nam, D.; Taitt, B. J.; Choi, K. Copper-based catalytic anodes to produce 2,5-furandicarboxylic acid, a biomass-derived alternative to terephthalic acid. ACS. Catal. 2018, 8, 1197-206.
27. Patil, S. K. R.; Lund, C. R. F. Formation and growth of humins via aldol addition and condensation during acid-catalyzed conversion of 5-hydroxymethylfurfural. Energy. Fuels. 2011, 25, 4745-55.
28. van, Z. I.; Wang, Y.; Rasrendra, C. B.; et al. Formation, molecular structure, and morphology of humins in biomass conversion: influence of feedstock and processing conditions. ChemSusChem 2013, 6, 1745-58.
29. Duan, J.; Chen, S.; Zhao, C. Ultrathin metal-organic framework array for efficient electrocatalytic water splitting. Nat. Commun. 2017, 8, 15341.
30. Indra, A.; Paik, U.; Song, T. Boosting electrochemical water oxidation with metal hydroxide carbonate templated prussian blue analogues. Angew. Chem. Int. Ed. Engl. 2018, 57, 1241-5.
31. Zhou, J.; Wang, Y.; Su, X.; et al. Electrochemically accessing ultrathin Co (oxy)-hydroxide nanosheets and operando identifying their active phase for the oxygen evolution reaction. Energy. Environ. Sci. 2019, 12, 739-46.
32. Zhou, B.; Li, Y.; Zou, Y.; et al. Platinum modulates redox properties and 5-hydroxymethylfurfural adsorption kinetics of Ni(OH)2 for biomass upgrading. Angew. Chem. Int. Ed. Engl. 2021, 60, 22908-14.
33. Zhuang, L.; Ge, L.; Yang, Y.; et al. Ultrathin iron-cobalt oxide nanosheets with abundant oxygen vacancies for the oxygen evolution reaction. Adv. Mater. 2017, 29, 1606793.
34. Song, F.; Hu, X. Exfoliation of layered double hydroxides for enhanced oxygen evolution catalysis. Nat. Commun. 2014, 5, 4477.
35. Das, T. K.; Jesionek, M.; Çelik, Y.; Poater, A. Catalytic polymer nanocomposites for environmental remediation of wastewater. Sci. Total. Environ. 2023, 901, 165772.
36. Chen, D.; Ding, Y.; Cao, X.; et al. Highly efficient biomass upgrading by a Ni-Cu electrocatalyst featuring passivation of water oxidation activity. Angew. Chem. Int. Ed. Engl. 2023, 62, e202309478.
37. Pang, X.; Bai, H.; Zhao, H.; Fan, W.; Shi, W. Efficient electrocatalytic oxidation of 5-hydroxymethylfurfural coupled with 4-nitrophenol hydrogenation in a water system. ACS. Catal. 2022, 12, 1545-57.
38. Woo, J.; Moon, B. C.; Lee, U.; et al. Collaborative electrochemical oxidation of the alcohol and aldehyde groups of 5-hydroxymethylfurfural by NiOOH and Cu(OH)2 for superior 2,5-furandicarboxylic acid production. ACS. Catal. 2022, 12, 4078-91.
39. Zhao, G.; Hai, G.; Zhou, P.; et al. Electrochemical oxidation of 5-hydroxymethylfurfural on CeO2-modified Co3O4 with regulated intermediate adsorption and promoted charge transfer. Adv. Funct. Mater. 2023, 33, 2213170.
40. Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. Condens. Matter. 1996, 54, 11169-86.
41. Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B. Condens. Matter. 1994, 49, 14251-69.
42. Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396.
43. Liechtenstein, A. I.; Anisimov, V. I. V. I.; Zaanen, J. Density-functional theory and strong interactions: orbital ordering in Mott-Hubbard insulators. Phys. Rev. B. Condens. Matter. 1995, 52, R5467-70.
44. Jamal, M.; Shahriyar, N. S.; Sharif, A. Effects of transition metal (Fe, Co & Ni) doping on structural, electronic and optical properties of CuO: DFT + U study. Chem. Phys. 2021, 545, 111160.
45. Heinz, U.; Song, H.; Chaudhuri, A. K. Dissipative hydrodynamics for viscous relativistic fluids. Phys. Rev. C. 2006, 73, 034904.
46. Wang, H.; Niu, C.; Liu, W.; Tao, S. d-Electron tuned CoMoP for enhance 5-hydroxymethylfurfural oxidation and HER. Appl. Catal. B. Environ. 2024, 340, 123249.
47. Wu, T.; Xu, Z.; Wang, X.; et al. Surface-confined self-reconstruction to sulfate-terminated ultrathin layers on NiMo3S4 toward biomass molecule electro-oxidation. Appl. Catal. B. Environ. 2023, 323, 122126.
48. Cai, X.; Peng, F.; Luo, X.; et al. Understanding the evolution of cobalt-based metal-organic frameworks in electrocatalysis for the oxygen evolution reaction. ChemSusChem 2021, 14, 3163-73.
49. Hu, L.; Hu, Y.; Liu, R.; Mao, Y.; Balogun, M. S.; Tong, Y. Co-based MOF-derived Co/CoN/Co2P ternary composite embedded in N- and P-doped carbon as bifunctional nanocatalysts for efficient overall water splitting. Int. J. Hydrogen. Energ. 2019, 44, 11402-10.
50. Lu, Y.; Liu, T.; Dong, C. L.; et al. Tailoring competitive adsorption sites by oxygen-vacancy on cobalt oxides to enhance the electrooxidation of biomass. Adv. Mater. 2022, 34, e2107185.
51. Li, S.; Wang, S.; Wang, Y.; et al. Doped Mn enhanced NiS electrooxidation performance of HMF into FDCA at industrial-level current density. Adv. Funct. Mater. 2023, 33, 2214488.
52. Ge, R.; Wang, Y.; Li, Z.; et al. Selective electrooxidation of biomass-derived alcohols to aldehydes in a neutral medium: promoted water dissociation over a nickel-oxide-supported ruthenium single-atom catalyst. Angew. Chem. Int. Ed. Engl. 2022, 61, e202200211.
53. Sun, Y.; Wang, J.; Qi, Y.; Li, W.; Wang, C. Efficient electrooxidation of 5-hydroxymethylfurfural using co-doped Ni3S2 catalyst: promising for H2 production under industrial-level current density. Adv. Sci. 2022, 9, e2200957.
54. Lu, L.; Wen, C.; Wang, H.; Li, Y.; Wu, J.; Wang, C. Tailoring the electron structure and substrate adsorption energy of Ni hydroxide via Co doping to enhance the electrooxidation of biomass-derived chemicals. J. Catal. 2023, 424, 1-8.
55. Moysiadou, A.; Lee, S.; Hsu, C. S.; Chen, H. M.; Hu, X. Mechanism of oxygen evolution catalyzed by cobalt oxyhydroxide: cobalt superoxide species as a key intermediate and dioxygen release as a rate-determining step. J. Am. Chem. Soc. 2020, 142, 11901-14.
56. Chen, Z.; Cai, L.; Yang, X.; et al. Reversible structural evolution of NiCoOxHy during the oxygen evolution reaction and identification of the catalytically active phase. ACS. Catal. 2018, 8, 1238-47.
57. Li, S.; Sun, X.; Yao, Z.; et al. Biomass valorization via paired electrosynthesis over vanadium nitride-based electrocatalysts. Adv. Funct. Mater. 2019, 29, 1904780.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].