

Electrochemical conversion of CO2 via C−X bond formation: recent progress and perspective

Abstract
With the depletion of traditional energy sources and growing environmental concerns, it is becoming increasingly urgent to develop green, low-emission renewable energy technologies to replace fossil fuel-driven methods that emit carbon dioxide (CO2). Currently, the electrochemical production of high-value-added chemicals and fuels from CO2 has aroused great interest from scientists. However, to make full use of CO2 for the preparation of chemicals, it is necessary to expand the range of electrosynthesis methods, in particular by expanding reaction pathways through the reaction of CO2 with different substrates. In general, CO2 can form new covalent bonds with substrate molecules through the formation of C−X bonds, including C−H, C−C, C−N, C−O, and C−S bonds, which would expand the range of possible products by diversifying the reaction pathway. In this review, we focus on the research progress in electrochemical conversion of CO2 through C−X bond formation. We start by examining fundamentals of the reactions and summarizing the reaction modes. Next, we discuss the electrosynthesis of C−X bonds (C−H, C−C, C−N, C−O, C−S) using CO2 and different substrate molecules. Finally, (i) strategies for the design and activity optimization of catalyst materials and (ii) the future development of forming five types of bonds from CO2 and small molecules are discussed, along with an outlook on their future research prospects.

Keywords
INTRODUCTION
Excessive emissions of carbon dioxide (CO2) have been recognized as one of the main causes of global warming. With the progress of society and the development of science and technology, increasing attention has been paid to the sustainable development of carbon energy to solve the contradiction between carbon energy and CO2 emissions. Aside from crafting strategies for mitigation and removal, CO2 conversion to produce useful fuels/chemicals using sustainable energy is a promising route to enable a carbon-neutral cycle in modern industrial society. In 2013, the concept of “green carbon science” was first put forward by Professor Mingyuan He, which is defined as “the study and optimization of the transformation of carbon-containing compounds and the relevant processes involved in the entire carbon cycle from carbon resource processing, carbon energy utilization, CO2 fixation, and carbon recycling to utilize carbon resources efficiently and minimize net CO2 emission”[1]. The design of many environmental catalytic systems and process technologies is guided by the general principles of this concept. To facilitate CO2 capture and conversion, to date, various strategies have been developed to use CO2 as a feedstock for chemicals and fuels, such as photochemical catalysis, thermochemical catalysis, and electrochemical catalysis, among others. Among these approaches, electrocatalysis can use renewable energy to catalytically synthesize different types of chemicals from the earth’s abundant renewable resources under mild conditions with high efficiency and selectivity[2-5], which is the key to future new energy storage and conversion technologies[6-10]. The activity and selectivity can be optimized by a variety of powerful tools, such as the control reaction pathways, the design of catalysts, the optimization of electrolytes, and the modulation of reaction potentials, which is expected to realize the energy and chemical de-fossilization, low carbon green, and distributed development[11-12].
Electrosynthesis, as an efficient and environmentally friendly synthesis method, employs electrons as redox reagents[13-14]. In CO2 fixation, coupling CO2 with small molecule compounds or organic compounds by electrochemical methods has recently been promoted as an environmentally friendly and sustainable synthesis technique[15-19]. In general, CO2 can form new covalent bonds with substrate molecules through the formation of C−X bonds, including C−H, C−C, C−N, C−O, and C−S bonds, which would enrich the reaction pathway and produce various chemicals [Figure 1]. During the electrosynthesis, the inert CO2 molecule must first be adsorbed and activated on the catalyst surface because CO2 is a stable molecule and breaking or hydrogenating one of the oxygen atoms of CO2 is needed for its utilization in electrochemical C−X bond formation reactions. The activated CO2 molecules subsequently combine with the X source to form intermediates. Through the transfer of electrons and protons, C−X bonds can be constructed to produce the final product. Among various reaction routes for CO2 conversion, the C−H bond is constructed from the hydrogenation of CO2 to produce CO, CH4, HCOOH, and C2+ products which have been intensively investigated. However, studies on the construction of C−C, C−N, C−O, and C−S bonds are still limited and achieving high efficiency and selectivity remains a great challenge. Therefore, designing efficient electrochemical systems for the formation of different C−X bonds is highly desired[20-21].
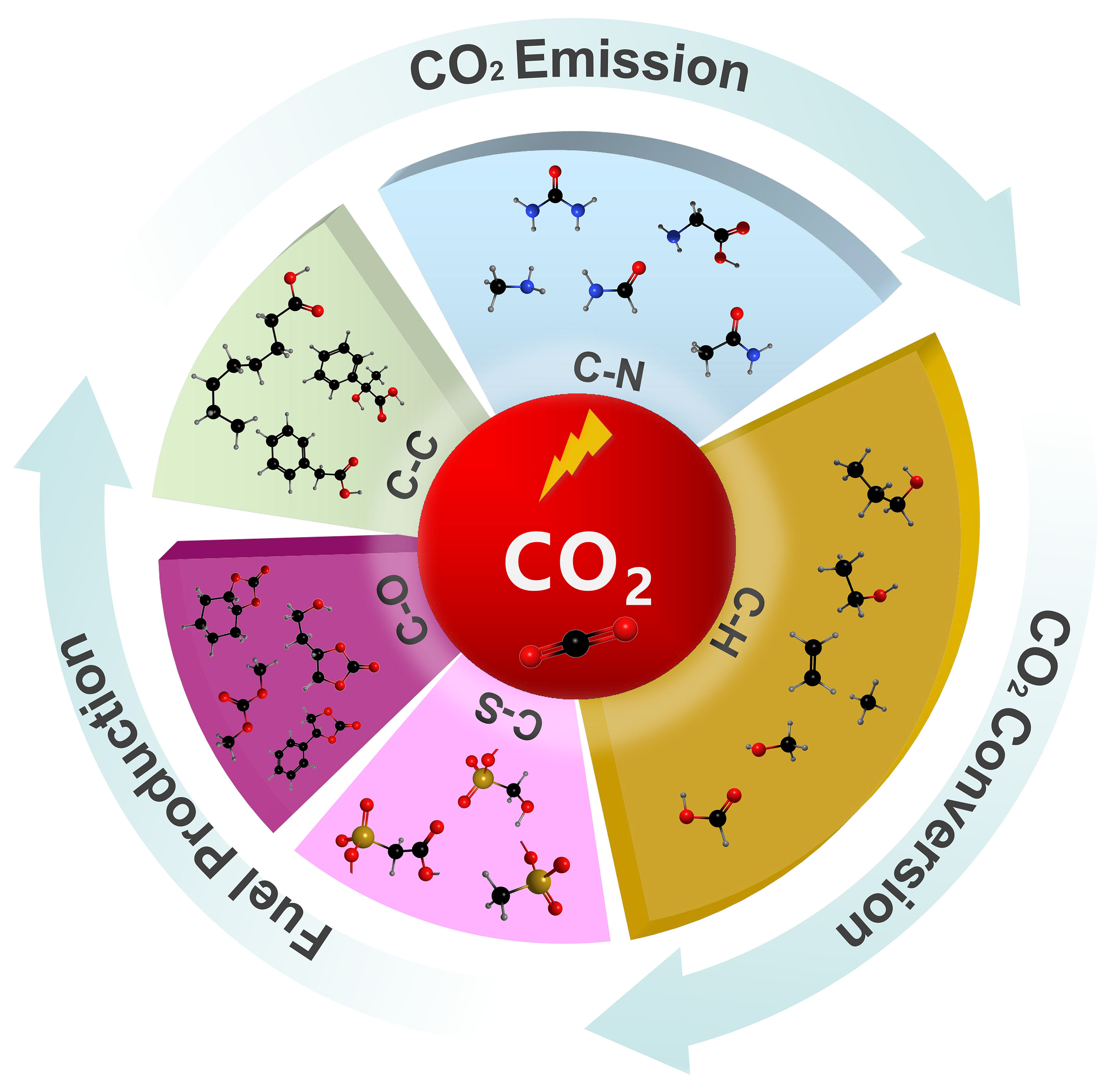
Figure 1. Sustainable fuel and chemical production through the formation of C−H, C−C, C−N, C−O, and C−S bonds.
This paper reviewed the recent advances in the electrosynthesis of C−X bonds (C−H, C−C, C−N, C−O, C−S) using CO2 and substrate molecules, including the electrochemical C−H bond formation for hydrocarbon/carbohydrate, electrochemical C−C bond formation for carboxylation of CO2, electrochemical C−N bond formation for organic nitrates synthesis, electrochemical C−O bond formation for cyclic carbonates and dimethyl carbonate (DMC) and electrochemical C−S bond formation for sulfonates[22-23]. Recent developments, prospects, and challenges in electrochemical CO2 utilization, and the fundamentals of electrocatalytic reactions on the initial activation of CO2 and product selectivity will also be described. Finally, improvement strategies for the shortcomings of electrocatalytic CO2 coupled organic reactions and outlooks on their future research prospects are proposed[24].
FUNDAMENTALS OF ELECTROCATALYTIC REACTIONS
The electrochemical CO2-catalyzed reaction is capable of using various substrate molecules to prepare advanced products in addition to the direct conversion of CO2 and water into carbon-containing chemicals[25-28]. Typically, Figure 2A and B can represent the construction of C−H bonds between water and CO2, which are reduced to a one-carbon product and a multi-carbon product, respectively. Figure 2C can represent the construction of C−N and C−S bonds between CO2 and N (S)-containing small-molecule compounds, and Figure 2D can represent the form of the reaction that forms C−C and C−O bonds.
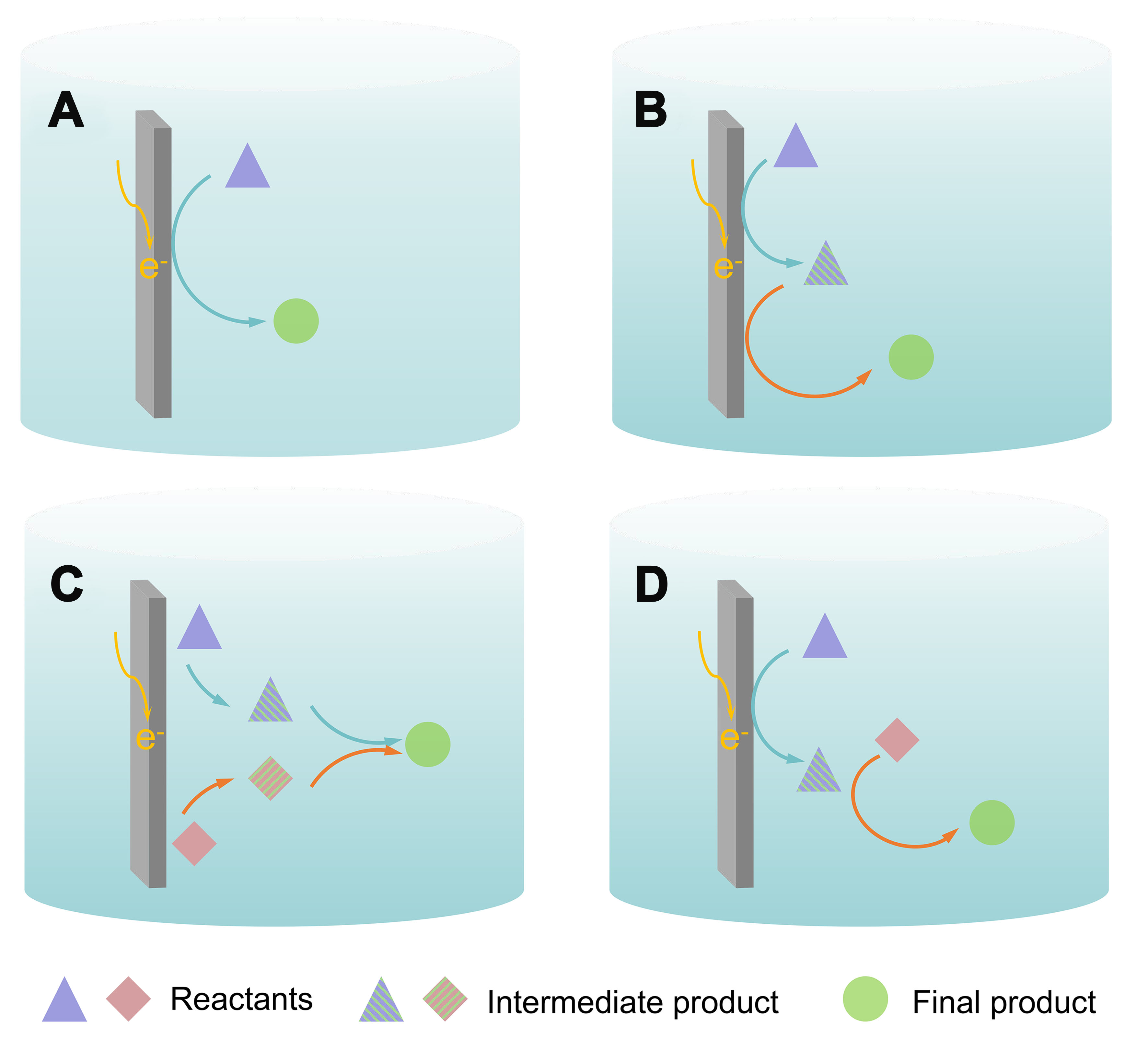
Figure 2. Different electrocatalytic reaction pathways. (A) Illustration reactions of a→d; (B) Illustration reactions of a→b→d; (C) Illustration reactions of a+c→d; (D) Illustration reactions of a→b, b+c→d. a and c: reactants; b: intermediate or intermediate product; d: final product.
According to the different electrocatalytic reaction pathways, there are generally two types of reaction modes.
One is the tandem reaction mode. The use of desorbed intermediates as local self-sufficient sources for following reactions is also of great research interest and value, which can be electrocatalytic or thermodynamically spontaneous. This tandem reaction concept has the obvious advantage of allowing more complex chemicals obtained by single-step electrocatalytic reactions and simplifying the synthesis process by avoiding the separation of intermediates, thus showing higher economic attractiveness. The concept of tandem reactions, as reviewed by Tang et al., can broaden the application of electron utilization to more complex chemicals that cannot be obtained by one-step electrocatalytic reactions[29]. Inspired by this, Peng et al. discovered that double sulfur vacancies on copper hexagonal sulfide serve as effective electrocatalytic centers, stabilizing CO* and OCCO* dimers and facilitating the coupling of CO−OCCO to produce C3 compounds [Figure 3A][30]. In another study of the CO2 electrocarboxylation reaction, ketones are first reduced by two-electron transfer at the cathode, and then further converted to carboxylates by coupling with CO2 [Figure 3B][31]. It is also a typical pathway for a tandem reaction.
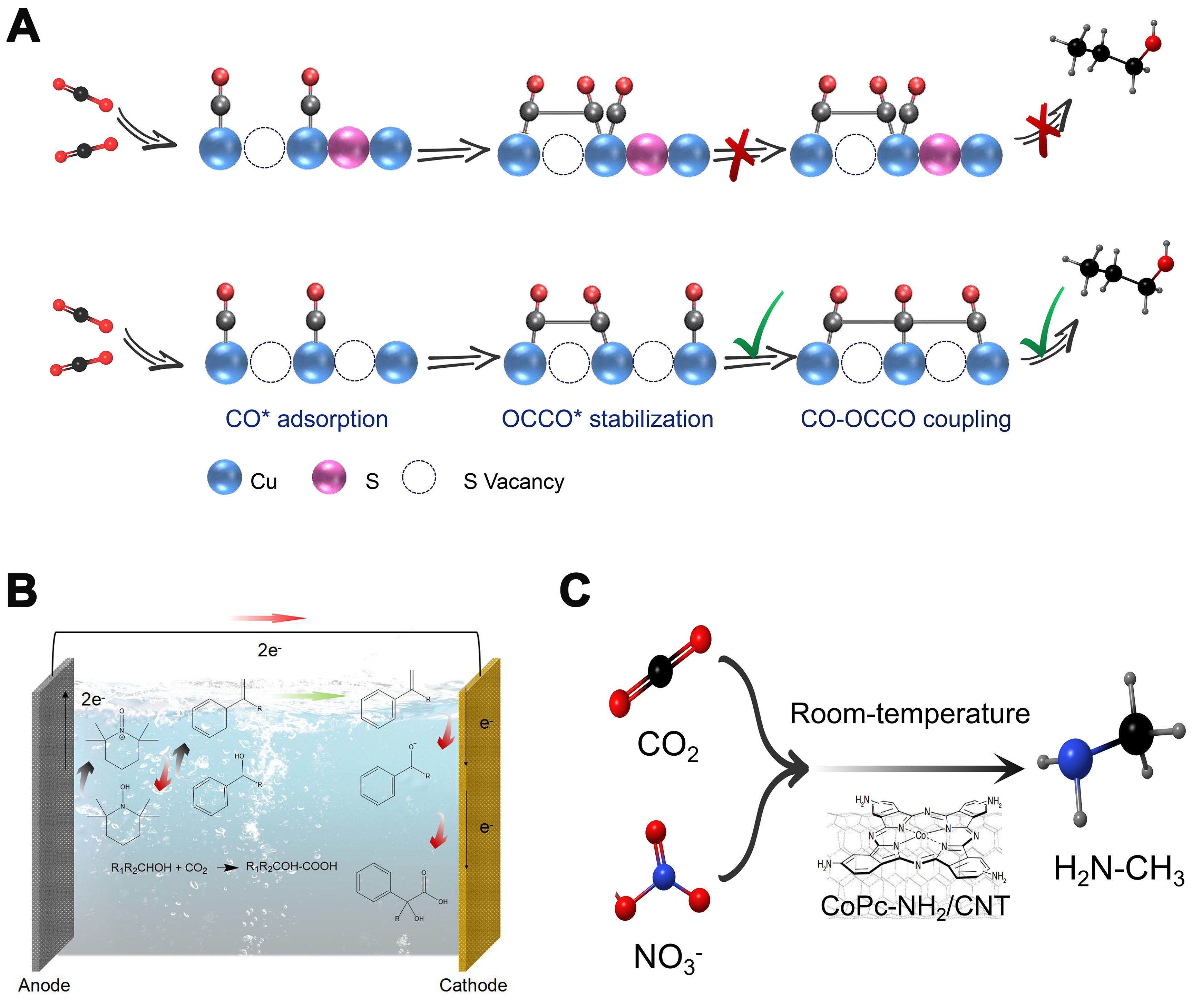
Figure 3. Typical tandem reaction and coupling reaction modes. (A) Mechanism of n-propanol formation on adjacent CuSx-DSV, showing the dimerization of CO−CO followed by CO−OCCO coupling; (B) Electrosynthesis of α-hydroxy acids through the electrocatalytic coupling of alcohols and CO2; (C) Cascade electrosynthesis of methylamine from CO2 and NO3-. CuSx-DSV: CuSx double sulfur vacancy.
The other is the coupling reaction mode. The electrocatalytic CO2 conversion reactions can potentially derive new chemistries by simultaneous activation of small molecules other than CO2 at the active site of the catalyst to increase activity and alter selectivity, which can be achieved by adjacent active reaction sites and coupling reactions[32-34]. Wu et al. recently proposed a possible coupling reaction pathway for the electrocatalytic synthesis of methylamine[35]. This pathway integrates the electrochemical reduction of CO2 (CO2RR) and nitrate (NO3-RR) [Figure 3C]. First, CO2RR and NO3-RR can occur independently to form formaldehyde (HCHO) and hydroxylamine (NH2OH) intermediates, respectively. Then, formaldoxime is produced by nucleophilic NH2OH attack on HCHO and then reduced to N-methyl hydroxylamine. Finally, methylamine is obtained by further reduction of N-methyl hydroxylamine. It is noteworthy that the key to the generation of the C−N bond is the condensation of NH2OH and HCHO intermediates from NO3-RR and CO2RR, respectively.
INTEGRATED ELECTROCATALYTIC REACTIONS
C−H bond formation
The generation of C−H bonds occurs during the CO2 reduction reaction (CO2RR) with multiple proton-electron transfer steps that yield carbon-based products. A typical CO2 electrolysis usually involves a multi-electron/proton transfer process: (i) CO2 adsorption and activation on electrolyte/electrode interface; (ii) additional electron/proton transfer to produce the desired product; and (iii) release of the product into the electrolyte. Therefore, both electrocatalysts and electrolyte are pivotal for designing efficient electrochemical devices[36-38]. Generally, various electrocatalysts have been utilized in CO2 electrolysis, ranging from homogeneous to heterogeneous catalysts. Homogeneous catalysts dissolved in the electrolyte, generally organic or organo-metallic complexes, such as metal pyridine, and metal porphyrins, usually promote CO2 reduction coupling by facilitating the transfer of electrons and protons[39]. In contrast to homogeneous electrocatalysts, heterogeneous electrocatalysts are more concerned. Electrocatalysts are typically solid materials, with the reaction predominantly taking place at the interface between the electrolyte and the catalyst. Up to date, various electrocatalysts (e.g., Au, Ag, Pd, Co, Bi, Cu, Fe, Sn, carbon-based materials,
As shown in Figure 4, electrochemical CO2RR involves the transfer of multiple protons and electrons, which involves diverse pathways. CO2 can undergo reduction to yield a variety of products comprising both single- and multiple-carbon compounds, including carbon monoxide (CO), methane (CH4), methanol (CH3OH), formate (HCOO-), ethylene (C2H4), ethanol (C2H5OH), and propanol (C3H7OH), among others. There are three types of bonding steps in the CO2RR route: oxygen hydrogenation, carbon hydrogenation, and carbon-carbon coupling[50-53]. Hydrocarbonation often occurs during the evolution of intermediates and the formation of products[54-56]. By calculating the relevant intermediates, the resulting hydrogen (H2), HCOO-, and hydrocarbon moieties can be distinguished[57]. The key factors affecting the distribution of these three products are the H* binding energy, the HCOO* binding energy, and possibly the *CO binding energy. The reduction potential of CO2 is typically more negative than the hydrogen evolution potential. At extremely negative potentials, the hydrogen evolution reaction (HER) tends to take precedence. Therefore, it is crucial to tailor catalyst characteristics to suppress the HER and enhance the multiple proton and electron transfer processes, enabling precise control over electrocatalysis for the targeted product.
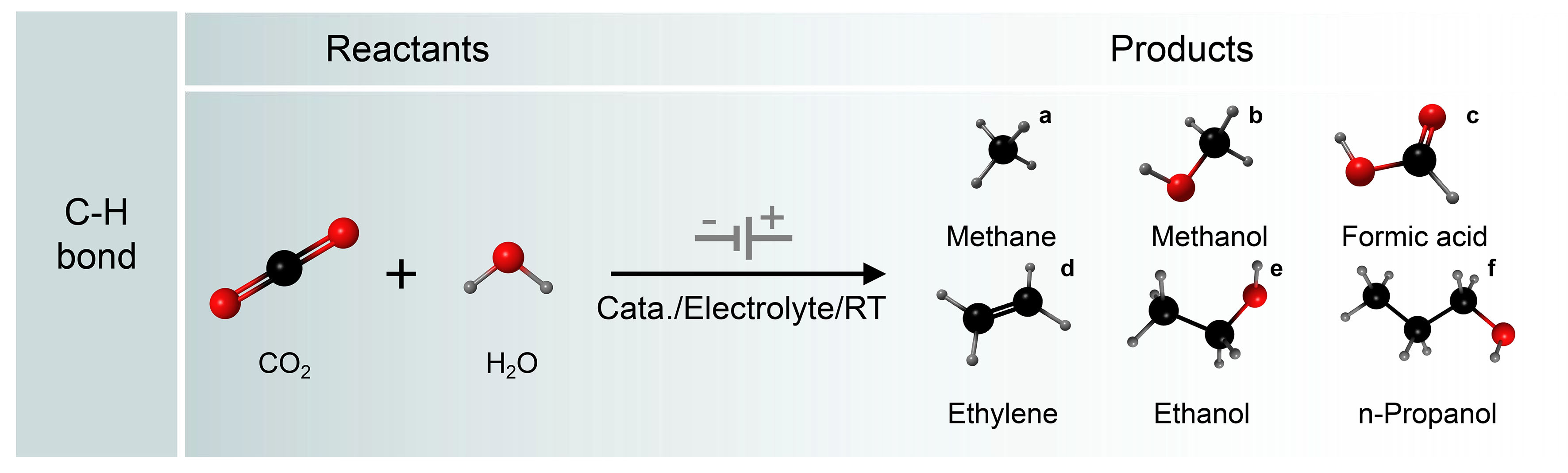
Figure 4. C−H bond formation by electrocatalytic reaction of CO2 and H2O.
Up to now, significant endeavors have been dedicated to enhancing the efficiency and specificity of CO2 reduction, ranging from the advancement of catalysts to the design of electrolytes and electrolyzers. Among them, designing robust electrocatalysts with self-supported nanostructures and desired features is very important to improve the catalytic ability of CO2RR.
Numerous reports have demonstrated the excellent catalytic properties and potential of carbon-based materials for CO2RR [Figure 5A][58]. However, the active sites of the catalysts have been the focus of debate. Currently, various forms of nitrogen species (e.g., pyridinic N, pyrrolic N, and graphitic N) doped in carbon materials, carbon atoms surrounding the nitrogen dopants, and carbon atoms with topological structural defects are considered to be the active sites for CO2RR. Therefore, to develop highly active carbon-based catalysts, it is necessary to identify the active sites of the catalysts. Kondo et al. suggested that in the CO2RR process, the carbon atoms adjacent to pyridinic N can act as Lewis base sites to adsorb CO2, which makes the reduced carbon atoms the active sites for CO2RR[59]. However, Guo et al. showed that the acidic CO2 molecules can only adsorb on the pyridinic N sites, and the Lewis base sites for CO2RR are generated by pyridinic N rather than graphitic N. Therefore, pyridinic N is considered as the active site for CO2RR[60].
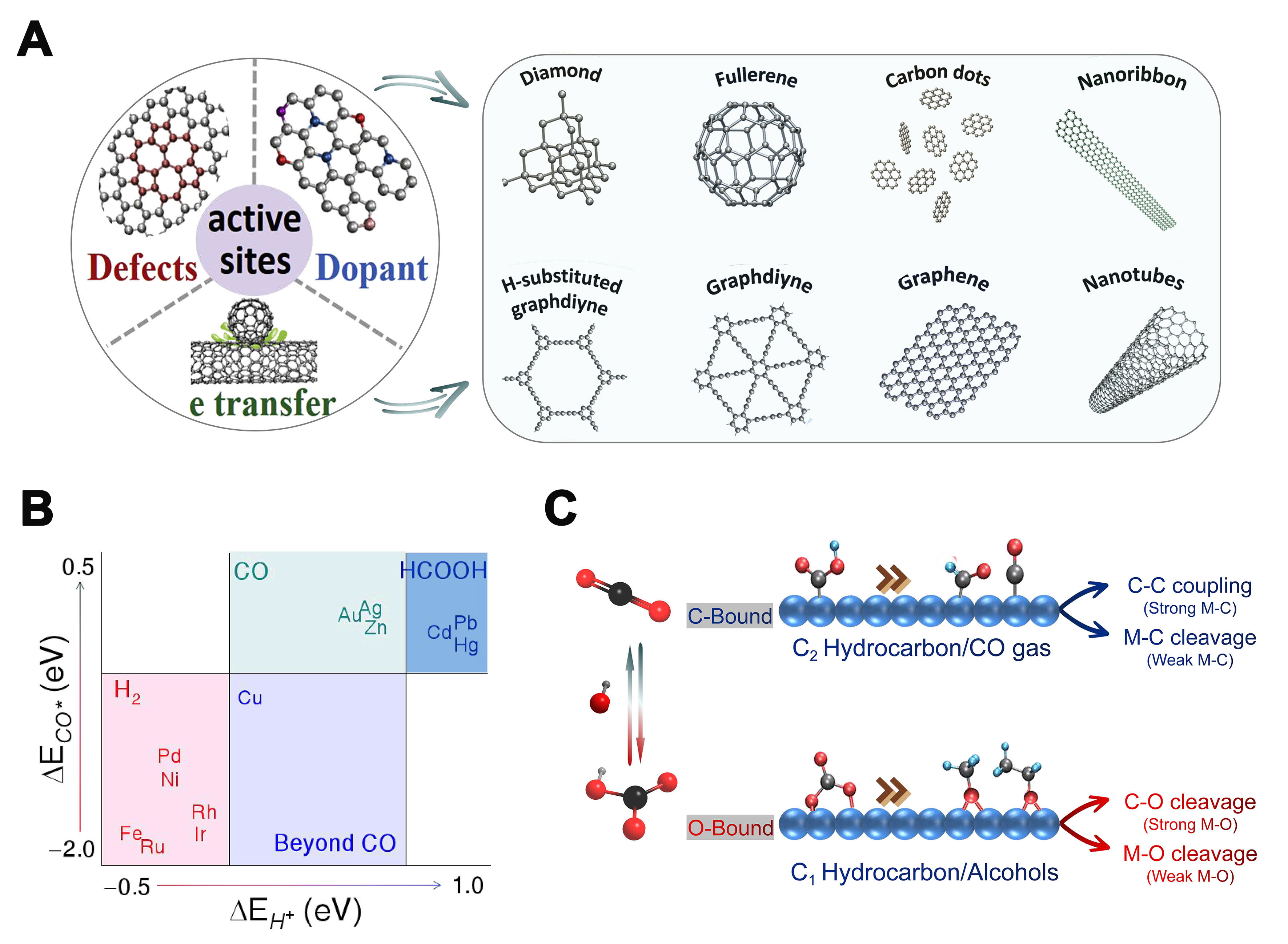
Figure 5. (A) Design of effective catalytical active sites in carbon-based metal-free electrocatalysts; (B) The adsorption energies of the CO and H intermediates on diverse metals and the predominant products of CO2RR on them; (C) The impact of surface-bound carbon and oxygen reaction intermediates on the selectivity of CO2RR products. CO2RR: CO2 reduction reaction.
Metal-based catalysts are often the most common electrocatalysts in the CO2RR. Typically, the diverse transition metals yield distinct primary products as a result of their varying affinities towards the intermediates *COOH and *CO. Bagger et al. classified more than ten metals through more detailed calculations of the adsorption energy correlations of species such as H*, *COOH, HCOOB*, and *CO[61]. The study found that: (1) Pt, Pd, and Ni have great adsorption energy on H* and *CO; thus, HER becomes the dominant one; (2) Metals such as Au and Ag, which have CO as the main product, have weak adsorption energy for H*, *COOH, and *CO, etc., and *CO has the opportunity to desorb from the surface to produce gas-phase CO products. At the same time, it is also their weak adsorption of intermediate *COOH that makes the production of CO on these metals require a certain overpotential. To reduce the overpotential (enhance *COOH adsorption) while maintaining high CO-producing activity (maintain *CO adsorption), measures are needed to break the linear relationship between *COOH and *CO adsorption energies[62-64]; (3) Sn, Pb, Hg, and other metals with formic acid as the main product tend to have the weakest adsorption of H*, while they also have weak adsorption of *CO and *COOH and moderate adsorption of HCOOB*; (4) Cu has relatively moderate adsorption of both H* and *CO which makes *CO neither too easily desorbed from the surface to produce gas-phase CO products, nor too strongly adsorbed on the surface to become toxic, but has the opportunity to be further reduced to hydrocarbons or polycarbonate products [Figure 5B].
Katayama et al. further clarified the surface reaction properties of metal catalysts [Figure 5C][62]. They deduced that when the metal has a propensity to bond with carbon rather than oxygen, intermediates such as *CO, *CHO, and *carboxylate are readily formed on the metal surface. In contrast, if the metal tends to bond with oxygen, the adsorption of carbonates in easily formed states occurs. The different binding types will affect the formation of C−H bonds, which, in turn, will lead to the eventual formation of CO, C2 products, etc. of intermediate species adsorbed on the carbon end, while intermediate species adsorbed on the oxygen end eventually form C1, alcohols, etc.
Recent advancements have also focused on achieving industrially significant performance by optimizing gas diffusion electrodes (GDEs) in flow reactor systems, in addition to investigating reaction pathways[65]. A gas diffusion layer (GDL) and a catalytic layer (CL) are the key components of a GDE. The porous architecture enhances the rapid transport of CO2 to the interface between the gas, electrolyte, and catalyst via the rear of the GDL. Polymers/binders are usually used to coat the powder electrocatalyst on the GDL as the catalyst layer. In this device, CO2 electrolysis occurs at the gas-liquid-solid three-phase interface.
In order to enhance the CO2RR activity under high current conditions, diverse strategies have been employed to design the gas diffusion assembly. In a representative study, de Arquer et al. integrated ionomer with catalyst particles to enhance the diffusion of CO2 to the active catalyst surface, resulting in CO2 electrolysis to ethylene at the current density of 1 A·cm-2[66]. The electrolysis was conducted in 7 M KOH electrolyte, leading to an ethylene partial current density of 1.3 A·cm-2 with a cathodic energy efficiency of 45%. The results show that the catalyst ionomer bulk heterojunction (CIBH) consists of a metal and an ultra-thin ionomer layer with hydrophobic and hydrophilic properties, which separates the transport of gas, ions, and electrons. Recently, Li et al. used quaternary ammonia poly(ether ether ketone) (QAPEEK)-containing carbonyl groups as a bifunctional electrolyte for CO2RR[67]. The results show that the ionomer has ionic conductivity, which can activate CO2 at the catalyst-electrolyte interface and promote the production of ethylene. At a cell voltage of 3.54 V, the partial current density of ethylene was 420 mA·cm-2 without any electrolyte consumption, which meets the requirements for industrial-scale production. In another study, Endrődi et al. developed a poly (aryl piperidinium)-derived anion exchange membrane (PiperION) with enhanced carbonate conductance for the CO2RR to CO[68]. Utilizing a PiperION membrane in a membrane electrode assembly (MEA) resulted in a current density of more than 1 A·cm-2 for CO production. These works provide theoretical guidance for the industrial application of CO2 as a substrate for electrochemical reactions.
As shown in Table 1, to facilitate a more intuitive comparison and understanding for the readers, we have summarized some representative CO2 reduction reactions on nanostructured catalysts.
C1-C3 compound (CH4, formate, CO, C2H4, EtOH, acetic acid, and n-propanol, etc.) production from CO2RR
| Substrate | Product | Electrode | Electrolyte | Current density/mA·cm-2 or potential/RHE | FE/% |
| CO2, H2O | CH4 | Cu/CeO2@C[69] | 1 M KOH | 172 | 80.3 |
| CO2, H2O | CH4 | Cu/CeO2[70] | 1 M KOH | 300 | 67 |
| CO2, H2O | CH4 | CuNCN-500[71] | 1 M KOH | 300 | 66.3 |
| CO2, H2O | CH4 | Cu SA/F-GDY[72] | 1 M KOH | 241 | 72.3 |
| CO2, H2O | CH4 | Cu SAs/GDY[73] | 1 M KOH | 247 | 81 |
| CO2, H2O | Formate | MIL-68(In)-NH2[74] | 0.5 M KHCO3 | 108 | 94.4 |
| CO2, H2O | Formate | TMP-CH3-MCOF[75] | 0.5 M KHCO3 | 33.5 | 95.6 |
| CO2, H2O | Formate | CPs@V[76] | 0.5 M KHCO3 | 6.87 | 90.1 |
| CO2, H2O | CO | Cu/In NWs[77] | 0.1 M KHCO3 | 4.2 | 93 |
| CO2, H2O | CO | Bimetallic Cu/In[78] | 0.1 M KHCO3 | 17 | 92 |
| CO2, H2O | CO | Zn1Ni4-ZIF-8[79] | 1.0 M KHCO3 | 57.9 | 96.5 |
| CO2, H2O | CO | Cu/Zn-CP-1-30[80] | 0.5 M [Bmim]PF6 / MeCN | 165.5 | 94.3 |
| CO2, H2O | C2H4 | Cu-[CF2]n-5-CP[64] | 0.1 M CsI | 23.8 | 67.3 |
| CO2, H2O | C2H4 | MOF CuPz2[81] | 0.1 MKHCO3 | 12.4 | 70.2 |
| CO2, H2O | C2H4 | Cu-dimethyl-pyrazole[82] | 0.1 M KCl | 10.0 | 70.7 |
| CO2, H2O | C2H4 | CuPOF-Bpy/Cu2O@CNT[83] | 0.5 M KHCO3 | 31.2 | 71.0 |
| CO2, H2O | C2H4 | CuO/Ni SAs[84] | 1.0 M KOH | 811.5 | 54.1 |
| CO2, H2O | EtOH | Cu/C-0.4[85] | 0.1 M KHCO3 | -0.7 V | 91 |
| CO2, H2O | EtOH | Cu-DS[86] | 0.1 M KHCO3 | 70 | 52 |
| CO2, H2O | EtOH | Cu2S1-x[87] | 0.5 M KHCO3 | -0.3 V | 73.3 |
| CO2, H2O | Acetic acid | PcCu-TFPN[88] | 0.1 M KHCO3 | 11.3 | 90.2 |
| CO2, H2O | Acetic acid | NDD/Si RA[89] | 0.5 M NaHCO3 | 0.54 | 77.3 |
| CO2, H2O | Acetic acid | Cu(I)/BN-C[90] | 25 mol% [Emim]BF4 and 75 mol% water | 11.1 | 80 |
| CO2, H2O | n-propanol | Cu2O/Cu[91] | 0.1 M KHCO3 | 6.8 | 12.1 |
C−C bond formation
As the key skeleton of organic compounds, C–C bonds are one of the most common chemical bonds in organic compounds[15-17,92]. Among the many products of CO2 conversion, carboxylic compounds are a very important class of compounds that are the basic structural units of many functional molecules, natural products, and biologically active substances, as well as important chemical products and industrial raw materials[93,94]. Therefore, the synthesis of carboxylic acids and their derivatives by electrocarboxylation of CO2 is of great importance [Figure 6]. In recent years, the method of CO2 carboxylation by the electrochemical drive has received increasing attention and has undergone development. Electrochemical synthesis of carboxylic compounds by constructing C−C bonds will provide a new way for the green and sustainable advancement of the chemical industry[14,95-98].
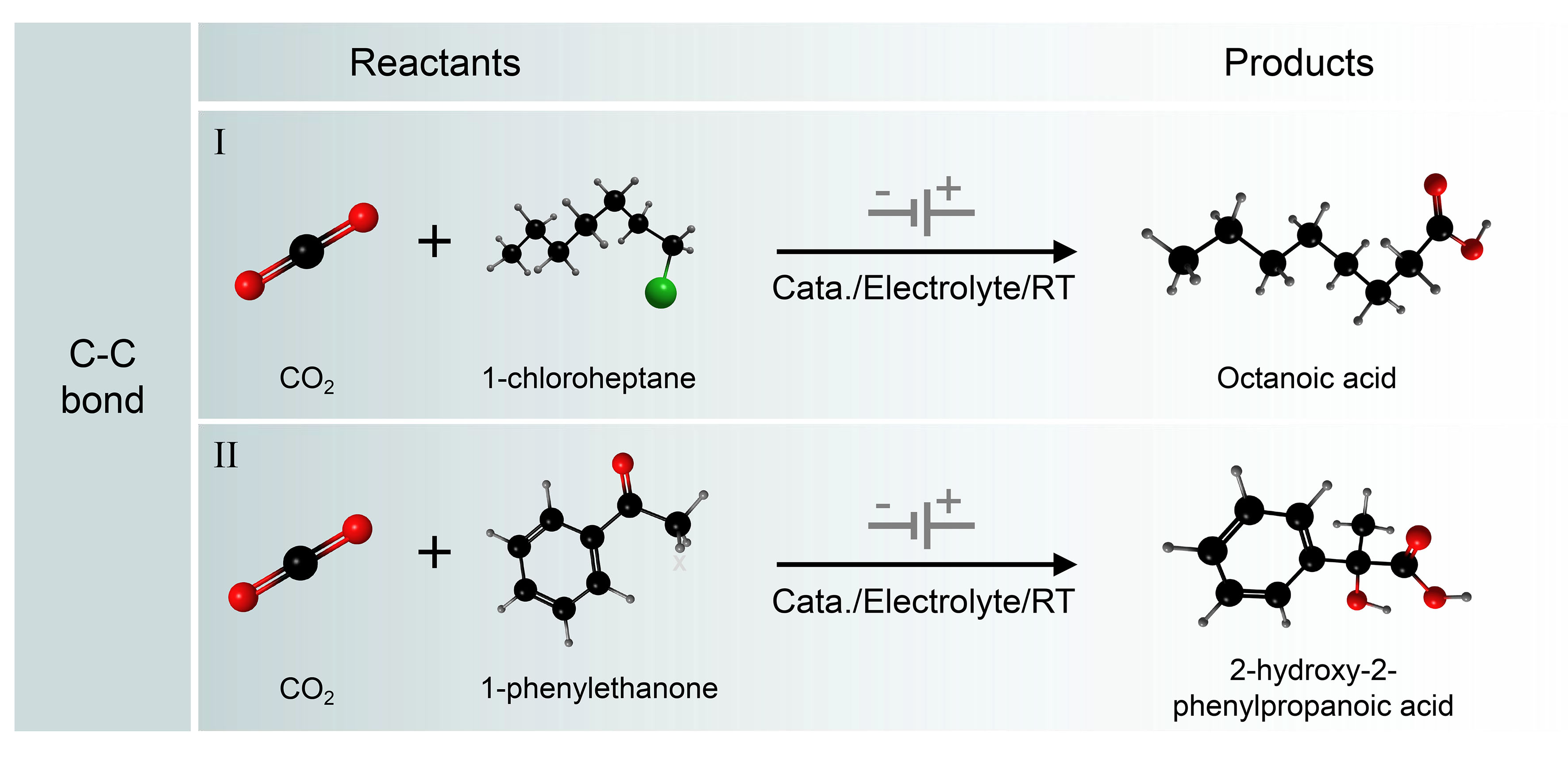
Figure 6. Electrocatalytic C−C coupling reactions. (I) Scope of aryl halides or alkyl halides; (II) Electrochemical CO2 fixation to unsaturated organic compounds.
In recent years, there has been a growing focus on research into electrocarboxylation, a process that utilizes CO2 as a feedstock. Under mild conditions, the carbon negative ions generated by the electroreduction of organic substrates can trap CO2 in the system, which, in turn, generates organic carboxylic compounds with multifunctional groups and higher added value[99-104]. For example, CO2 can react with olefins or alkynes to generate organic carboxylic acids through electroreductive addition reactions[99,100]. Aromatic ketones, whose C=O double bonds are conjugated with aromatic rings, are more likely to gain electrons on the electrode surface for electroreduction and carboxylation reaction[101,102]. On silver electrodes, organic halides can undergo electrocarboxylation reactions with CO2 to generate carboxylic acid compounds[103]. In addition, aromatic imine compounds can also react with CO2 to form carboxylic acid compounds, but the yields of such electrocarboxylation reactions tend to be low because they are inherently less stable and easily consumed by hydrolysis[104].
Generally, in a CO2 carboxylation reaction, the substrate usually carries polar covalent bonds or unsaturated patterns such as C−C double bonds, C−C triple bonds, carbonyl groups, imine patterns, and aromatic rings [Figure 7A]. The electrochemical carboxylation reaction with CO2 generally proceeds via the following two possible pathways [Figure 7B]. The first pathway is the direct electroreduction of CO2 to form a CO2 radical anion followed by a subsequent conversion to an acid [Figure 7C]. As a typical example, Zhang et al. reported that the unactivated skip dienes could be electrochemically carboxylated with CO2 to obtain valuable dicarboxylic acids. Control experiments and theoretical studies indicate that CO2 first undergoes a single electron transfer (SET) reaction to be reduced to its radical anion, followed by a slow radical addition reaction with the unactivated alkenes. Finally, the unstable alkyl radical is reduced to carbanions with nucleophilic properties, which are then coupled with CO2 to give the desired product[16]. The other pathway is the electroreduction of the substrate to form a carbon ion or radical anion, followed by a carboxylation reaction with CO2 [Figure 7D]. In this pathway, N-heteroarenes and organic halides are also an ideal class of carboxyl substrates in the presence of an electric current or a metal catalyst. Using CO2 as a carboxyl source, Sun et al. have achieved site-selective C−H bond carboxylation of pyridine and the related N-heteroarenes by switching different electrochemical cells[98]. In a divided electrochemical cell, C5-carboxylation occurs, whereas in an undivided electrochemical cell, C4-carboxylation occurs [Figure 7D (i)]. This method has good substrate applicability and functional group tolerance, providing a new approach for the preparation of important nitrogen-containing heterocyclic carboxylic acid compounds. In addition, Sun et al. report a versatile and practical electro-reductive Ni-catalytic system for the electrocatalytic carboxylation of unactivated aryl chlorides and alkyl bromides with CO2 [Figure 7D (ii)][96]. A variety of unactivated aryl bromides, iodides and sulfonates also perform this reaction without difficulty. Remarkably, they have also achieved a catalytic electrochemical carboxylation of aryl (pseudo)halides with CO2, avoiding the use of sacrificial electrodes. Mechanistic investigations suggest that the reaction might proceed via oxidative addition of aryl halides to Ni(0) complex, the reduction of aryl-Ni(II) adduct to the Ni(I) species, and subsequent carboxylation with CO2.
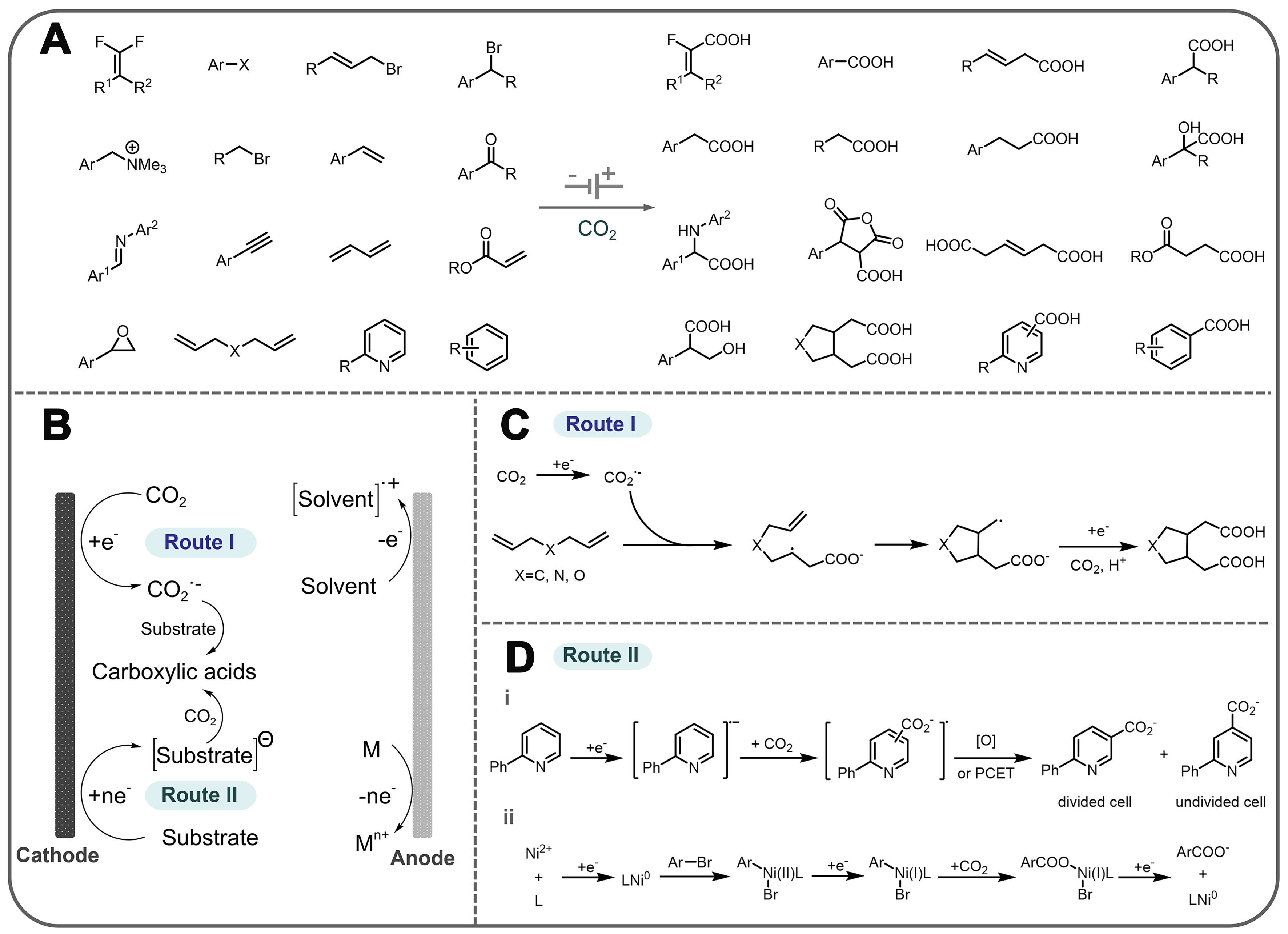
Figure 7. (A) List of electrochemical CO2 carboxylation reactions involving different types of substrates; (B) A comprehensive method for electrochemically catalyzed carboxylation reactions utilizing CO2 as a substrate to produce carboxylic acids; (C) The process of electrochemically-driven dicarboxylation of unactivated skipped dienes with CO2; (D) Mechanism of electrochemical carboxylation of (i) N-heteroarenes and (ii) aryl halide.
Despite many advances made in the construction of C−C bonds, there are still many problems that need to be further studied and solved in CO2 carboxylation reactions. On the one hand, there are side reactions such as electrochemical hydrogenation and electrochemical dimerization in the process of electrocarboxylation, which reduce the selectivity of the target products, and the current research generally suffers from the carboxylation product yield. On the other hand, many of the reactions require the use of co-catalysts, which complicates the electrochemical reaction system and is not conducive to the separation and purification of the products. Meanwhile, this reaction is typically conducted in non-aqueous organic solvents, and the evaporation of significant quantities of these solvents also leads to environmental pollution and substantial waste. Therefore, it is of great significance to design cheap and effective electrocatalytic materials and green catalytic systems to improve the reaction efficiency.
As shown in Table 2, we have summarized some representative examples of CO2 and substrate electrolysis through the construction of C−C bonds, and provided a detailed comparison of relevant parameters such as catalysts and yields among different reactions.
Selected examples of electrolytic conversion of CO2 and substrate by constructing C−C bonds
| Substrate | Electrode | Catalyst | Electrolyte | Solvent | Current or voltage | Yield |
 | (-)Pt-Ni(+)[105] | / | TBAI | DMF | 8 mA | 75% |
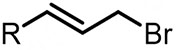 | (-)Ni-Mg(+)[106] | Co(OAc)2 PPh3 | TBAPF6 | DMF | 10 mA | 59% |
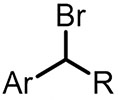 | (-)Ag NPs-Mg(+)[103] | / | TEAI | MeCN | 5 mA | 98% |
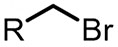
 | (-)C-Pt(+)[107] | / | TBABF4 | DMF | 4.5 V | 70% |
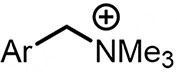
 | (-)C-Zn(+)[96] | NiBr2 DME dmbpy | LiClO4 | NMP | 8 mA | 70% |
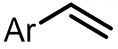 | (-)C-C(+)[92] | / | TEAI | DMF | 10 V | 70% |
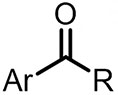 | (-)SS-Mg(+)[101] | / | TEAI | MeCN | 1.2 mA | 41% |
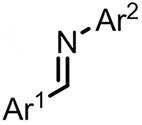 | (-)Ni-Al(+)[104] | / | TBAB | DMF | 10 mA | 68% |
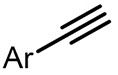 | (-)Ni-Al(+)[108] | / | TBAB | DMF | 10 mA | 70% |
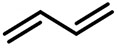 | (-)Ni-Al(+)[100] | / | TBAB | DMF | 10 mA | 84% |
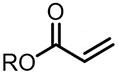 | (-)C-C(+)[93] | / | TEAI | DMF | 10 V | 76% |
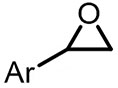 | (-)Pt-Mg(+)[17] | / | TBAI | DMF | 10 mA | 94% |
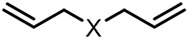 | (-)Ni-Al(+)[16] | / | TBAI | NMP | 12.5 mA | 78% |
 | (-)Fe-Zn(+)[98] | Cu(OTf)2 | TEAI | NMP | 5 mA | 72% (C5) |
| (-)Fe-Pt(+)[98] | / | TEAI | NMP | 6 mA | 85% (C4) | |
 | (-)GF-GF(+)[15] | / | TEAI | DMF | 20 mA | 87% |
 | (-)Nb-Zn(+)[95] | / | TBABF4 | NMP | 15 mA | 82% |
C−N bond formation
Compounds containing nitrogen have been applied extensively in chemical synthesis, medicinal chemistry, agriculture, and aerospace industries. Therefore, the development of efficient electrocatalytic C–N coupling strategies is of great significance for the production of organic nitrogen compounds. The combination of electrochemical CO2 with N2 reduction reaction (NRR), which utilizes the earth's abundant small molecules N molecules (N2, NO, NO2-, NO3-, etc.), is a viable approach to obtain valuable organic nitrogen compounds[109-111]. Electrocatalytic synthesis of nitrogen-containing chemicals can utilize green energy by decomposing water to provide protons under mild conditions of ambient temperature and pressure. It involves the adsorption, activation, and dissociation of inexpensive CO2 and nitrogen-containing compounds, facilitating the construction of C−N bonds and the generation and desorption of urea molecules. It is one of the viable alternatives to conventional processes with considerable economic viability and environmentally friendly sustainability. However, the limitation of this technology resides in the competition between the C−N coupling reaction and the reduction reactions of CO2 and N-containing small molecules, as well as HER. Driving both electrochemical reduction reactions (CO2RR/NRR) in the same electrochemical system to selectively produce the C−N coupling products is more difficult ] [Figure 8][112-115]. However, this approach offers significant advantages over currently reported industrial synthetic routes[116-118].
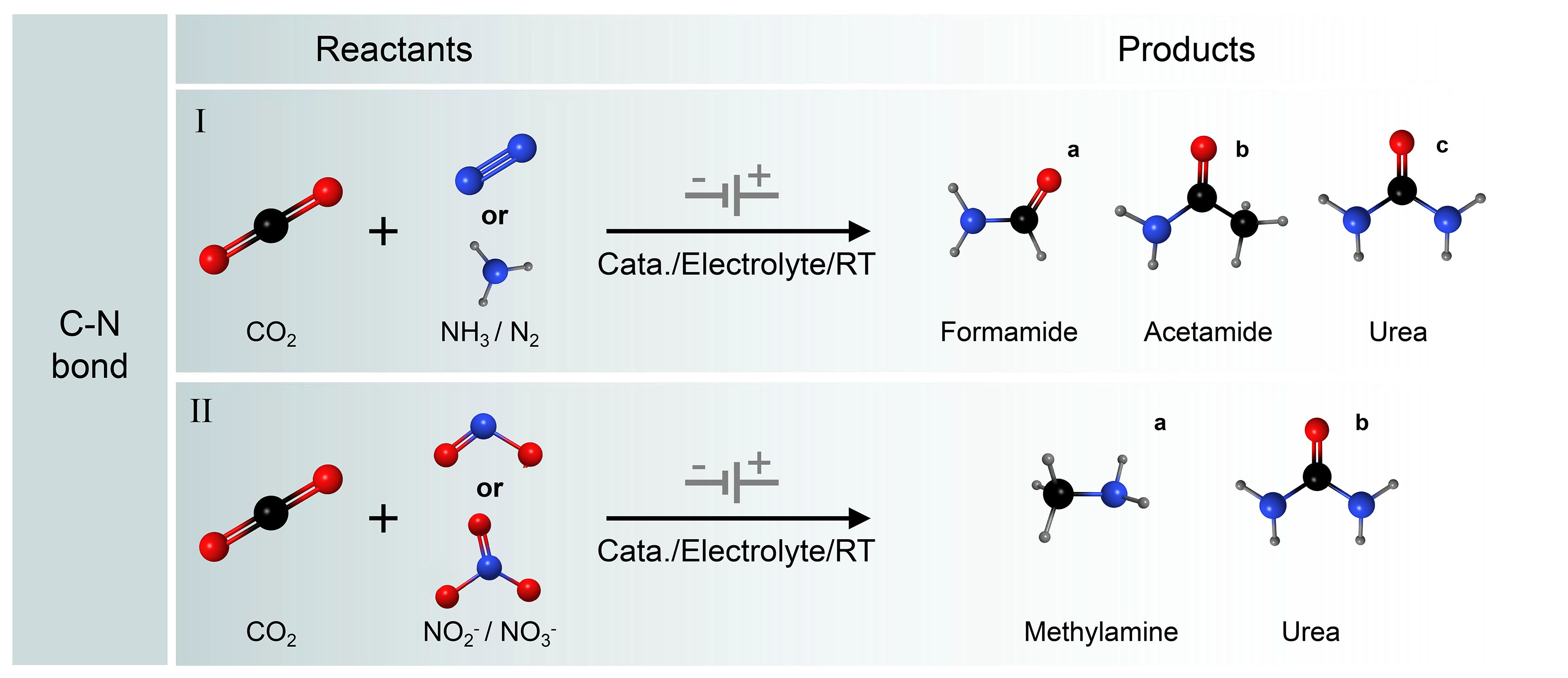
Figure 8. Electrocatalytic C−N coupling reactions. (I) Simultaneous reduction of CO2 and NH3/N2 to formamide/acetamide/urea; (II) Simultaneous reduction of CO2 and NO3-/NO2- to methylamine/urea.
To address the problem of C−N bond co-activation, Chen et al. pioneered a new method to prepare urea by electrochemically coupling CO2 and N2 in water. The electrocatalyst used in this method consists of PdCu alloy nanoparticles attached to TiO2 nanosheets with oxygen-rich vacancies. The yield of urea was
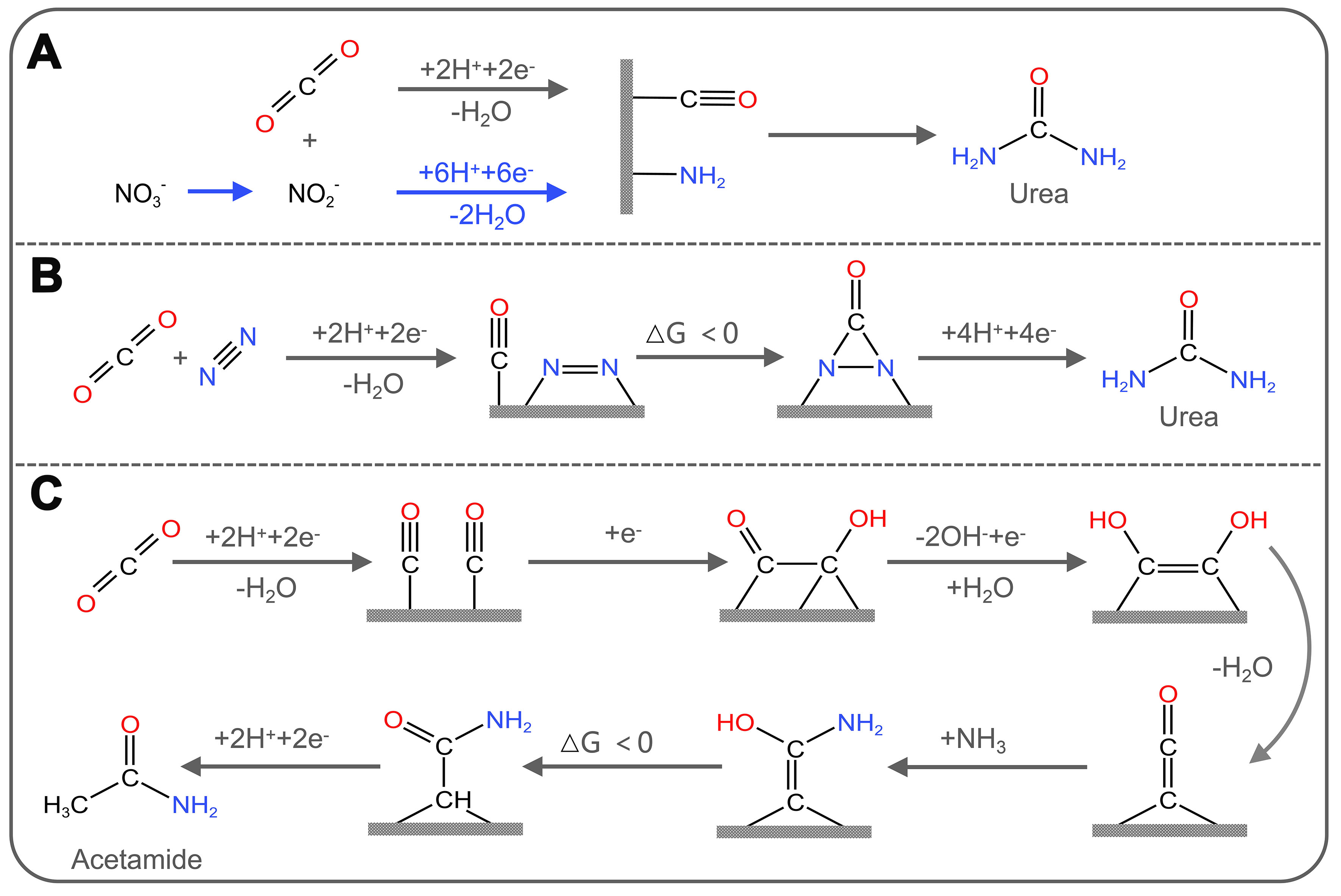
Figure 9. (A) Electrochemical synthetic pathway for urea from CO2 and NO3-/NO2-; (B) Electrochemical synthetic pathway for urea from CO2 and N2; (C) Electrochemical synthetic pathway for urea from CO2 and NH3.
Recently, remarkable progress has been made in C−N coupling reactions by electrocatalytic method using CO2 and NOx- as feedstocks. NOx- have relatively low dissociation energies and high aqueous solubility compared to non-polar N2, which makes them more suitable for the electrosynthesis of compounds containing nitrogen (e.g., urea, amides, amino acids, etc.) via C−N coupling reactions. Jouny et al. reported that the ketene intermediate (*C=C=O) produced by the coupling of two CO molecules adsorbed on the cathode acts as an electrophilic carbon center and is attacked by a nucleophilic amine to form the end product acetamide through successive protonation [Figure 9C][111]. Lv et al. achieved electrochemical coupling of NO3- with CO2 over In(OH)3. The selectivity and yield of urea were 53.4% and 533.1 μg·h-1·mg-1, respectively[125]. In another study, Xu et al. achieved a yield urea of 436.9 mmol·g-1·h-1 with a FE up to 66.4%, and an ultra-long cyclic stability of 1,000 h by modulating the C− and N− reduction kinetics through Cu doping and Pd4Cu1/FeNi(OH)2 interface[126]. In terms of amide product synthesis, Guo et al. reported a sustainable electrosynthesis method to synthesize formamide using CO2-derived HCOO- and NO2-[127]. The FE of formamide on low-coordinated copper nanocubes (ER-Cu) could reach 29.7%. Furthermore, using CO2-derived CH3OH and N2-derived NH3 as feedstocks for direct synthesis of formamide by electrooxidation is also an effective strategy. The FE of formamide was up to 41.2% on commercial boron-doped diamond (BDD) catalysts[128]. In terms of amino acid preparation, Fang et al. first discovered that the synthesis of C3+ amino acids from CO2 and NH3 can be electrocatalytically catalyzed by employing chiral Cu thin films (CCFs) as the electrodes, in which the FE of serine reached 1.2%[129].
These studies suggest that typically, the amalgamation of electrophilic intermediates-containing carbon with nucleophilic intermediates-containing nitrogen generated through the co-reduction of CO2 and nitrogen oxides is a pivotal process in the establishment of C−N bonds. Consequently, the strategic development of electrocatalysts featuring dual active sites is crucial for enhancing the selectivity of the C−N coupling reaction.
As shown in Table 3, we summarized some representative examples of CO2 electrolytic conversion and reactions with nitrogen-containing small molecules by constructing C−N bonds.
Selected examples of electrolytic conversion of CO2 and (NO, NO2-, NO3-, NH3, and N2)
| Substrate | Product | Electrode | Electrolyte | Current density or potential (vs. RHE) | FE |
| CO2, NO | Urea | Zn NBs[122] | 0.2 M KHCO3 | - 0.92 V | 11.3% |
| CO2, NO2- | Urea | ZnO-V[117] | 0.2 M NaHCO3 and 0.1 M NaNO2 | - 0.79 V | 23.3% |
| CO2, NO3- | Urea | In(OH)3-S[125] | 0.1 M KNO3 | - 0.6 V | 53.4% |
| CO2, NO3- | Urea | Pd4Cu1/FeNi(OH)2[130] | 0.1 M KHCO3 , 0.1 M KNO3 | - 0.5 V | 66.4% |
| CO2, N2 | Urea | Bi-BiVO4[110] | KHCO3 | - 0.40 V | 12.6% |
| CO2, N2 | Urea | BiFeO3/BiVO4[124] | 0.1 M KHCO3 | - 0.4 V | 17.2% |
| CO2, N2 | Urea | InOOH-100[115] | 0.1 M KHCO3 | - 0.40 V | 21.0% |
| CO2, N2 | Urea | VN-Cu3N-300[123] | 0.1 M KHCO3 | - 0.4 V | 28.7% |
| CO2, N2 | Urea | MOF Co-PMDA-2-mbIM[114] | 0.1 M KHCO3 | - 0.50 V | 49.0% |
| CO2, N2 | Urea | Ga79Cu11Mo10@C[113] | 0.1 M KHCO3 | - 0.40 V | 60.6% |
| CO2, NO3- | CH3NH2 | CoPc-NH2/CNT[31] | 0.1 M KNO3 | - 0.92 V | 13.0% |
| CO2, NH3 | Acetamide | Cu NPs[112] | 1 M KOH | - 0.58 V | 10.0% |
| CO, NH3 | Acetamide | Cu NPs[111] | 1 M KOH | 300 mA·cm-2 | 40.0% |
| CH3OH, NH3 | Formamide | BDD[128] | 0.5 M NaHCO3 | 120 mA·cm-2 | 41.2 % |
C−O bond formation
Redox-neutral CO2 conversion is an important process for producing various carbonates [Figure 10][131-135]. Among the synthesizable carbonates, DMC has a wide range of applications, often as a solvent for lithium batteries, reagents for biodiesel production, fuel additives, and intermediates for polycarbonate synthesis. The market size for DMC is expected to grow to $1.207 billion by 2025[136-138]. Thermochemical methods have been developed to synthesize DMC from CO2, but their inefficiencies and complex separation processes make them less likely to be industrialized. Therefore, the development of green and efficient synthesis methods remains an important challenge. To address this, Lee et al. explored a redox-neutral reaction approach to the formation of DMC from CO2[139]. A scheme for the redox-neutral electrochemical synthesis of DMC from CO2 and CH3OH is shown in Figure 11A. With the cathode and anode acting together in an undivided cell, solution-based non-homogeneous redox cycles are coupled to achieve electrical neutrality. Thus, electrons are transferred between the two electrodes by a CO2RR reaction at the cathode and two redox cycles at the anode: CO2/CO, Pd(0)/Pd(II), and Br-/Br2. Experimental results show a maximum FE of 60% for DMC production at room temperature. The mechanism study shows that CO2 and CH3OH may be reduced to produce CO and MeO- at the cathode. The Br2 generated by anodizing Br- oxidizes the Pd(0) complex to the Pd(II)Br2 complex, which can react with CO to produce the Pd(II)(CO)Br adduct. Finally, DMC was formed by the reaction of Pd(II)(CO)Br adduct with MeO- with generating Pd(0) complexes.
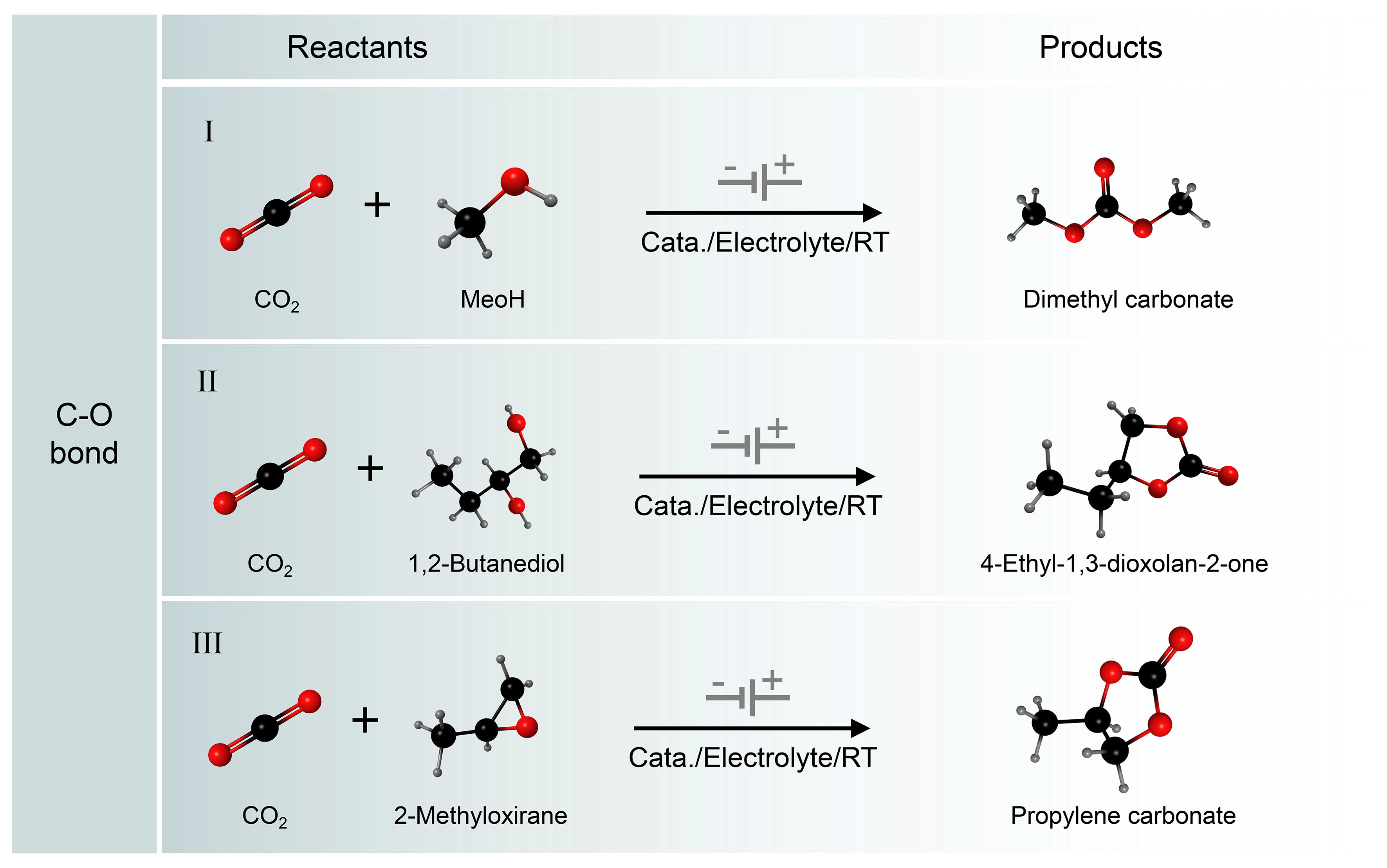
Figure 10. Electrocatalytic C−O coupling reactions. (I) Electrochemical conversion of CO2 to dimethyl carbonate without altering the redox state; (II) Electrosynthesis of Cyclic Carbonates from CO2 and Diols; (III) Electrosynthesis of cyclic carbonates from CO2 and epoxides.
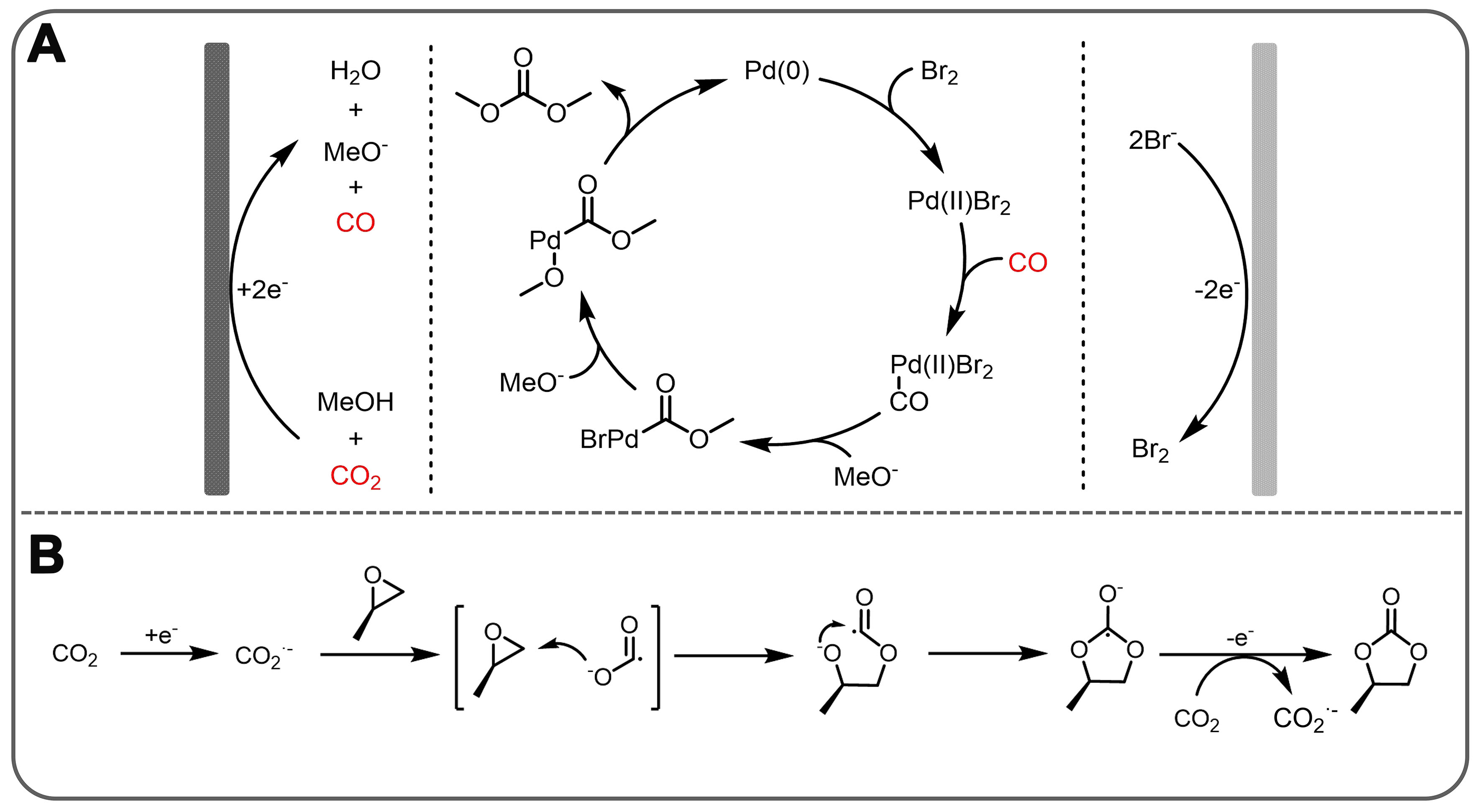
Figure 11. (A) Redox-neutral electrochemical system for dimethyl carbonate synthesis from CO2 and schematic of the proposed reaction mechanism for DMC synthesis; (B) Electrochemical synthetic pathway for propylene carbonate from CO2 and propylene oxide. DMC: Dimethyl carbonate.
Recently, Li et al. developed a novel Ni single-atom catalyst (SAC) featuring a dual-channel superstructure. This SAC exhibits a distinctive site coordination configuration, characterized by bonding with one axial oxygen atom and four planar nitrogen atoms[140]. The unique active sites are beneficial for the activation and conversion of CO2 to CO with a high FE of 99%. Furthermore, the preparation of CO2 to DMC with a purity of up to 80% was achieved through the petticoated Ni SAC. Studies on the mechanism have demonstrated that the axial oxygen coordination arrangement of the catalyst expedites the convergent paired electrosynthesis by diminishing the energy barriers associated with the formation of the *COOH intermediate and the dissociation of H2O and CH3OH.
The coupling of CO2 with epoxides with the formation of cyclic carbonates is a highly attractive 100% atom economic reaction. It represents a greener and safer alternative to the conventional synthesis of cyclic carbonates from diols and toxic phosgene. The electrochemical synthesis of cyclic carbonates from diols and CO2 in a reusable ionic liquid (IL)-based electrolyte without additional catalyst was reported by Wang et al. The reaction of CO2·- with 1,2-butanediol is supposed to produce intermediates [Figure 10]. Then, the intermediate is converted into a linear carbonate during the esterification process, which immediately becomes a cyclic carbonate[141]. Xiao et al. concurrently pioneered a one-step synthesis of enantiomerically pure cyclic carbonates through the electrolysis of chiral epoxides and CO2 in an undivided cell. They utilized tetraethyl ammonium iodide (TEAI) in acetonitrile (MeCN) as the supporting electrolyte and solvent, maintaining a constant current [Figure 11B][142,143]. In another work, Pérez-Gallent et al. studied the reaction process of propylene oxide and CO2 to synthesize propylene carbonate on copper electrodes in detail[144]. The results indicated that the key to this reaction was the reduction of CO2 to CO2·- and the C−O bond was generated by CO2·- attacking the epoxide.
As shown in Table 4, we summarized some representative reactions of coupling CO2 and organic small molecule substrates by constructing C−O bonds.
Selected examples of electrolytic conversion of CO2 and (methanol, diols, epoxides, and propylene oxide)
| Substrate | Product | Electrode | Catalyst | Electrolyte | Solvent | Current or voltage | Yield |
| CO2, methanol | DMC | Au plate[139] | Pd/C NaBr | 0.1 M NaBr | Methanol | 12 mA·cm-2 | 60% |
| CO2, methanol | DMC | Ni Sas/OMMNC[140] | Pd/C KBr | 0.1 M KBr | Methanol | 12 mA·cm-2 | 80% |
| CO2, diols | Cyclic carbonate | (-)Ni-Mg(+)[141] | / | C4MIMBF4 | C4MIMBF4 | -1.7 V | 12% |
| CO2, epoxides | Cyclic carbonate | (-)SS-Mg(+)[142] | / | TEAI | MeCN | 2.88 mA | 54% |
| CO2, propylene oxide | Cyclic carbonate | Cu/CS-II[143] | / | TEAI | MeCN | 4.14 mA | 95% |
C−S bond formation
The construction of C−S bonds from CO2 and S-containing species is important to the synthesis of sulfonates, which is widely used in pharmaceuticals, agriculture, and biology industries. Inspired by the electrochemical C−N bond formation from CO2 and small-molecule N-sources, the C−S coupling may be in principle according to a similar reaction pathway, because S and N have similar properties from diagonal relationships [Figure 12]. Recently, Li et al. first constructed C−S bonds from CO2 and SO32- as the C and S resources on Cu-based catalysts[145]. The electrosynthesis was carried out in a modified gas diffusion half-cell configuration, where CO2 was supplied through the gas phase and 1 mol/L KOH with 200 mmol/L SO32- was used as the electrolyte. After postreaction electrolysis at -0.68 V [vs. the reversible hydrogen electrode (RHE)] for 1 h, hydroxymethanesulfonate (HMS), sulfoacetate (SA), and methanesulfonate (MS) products were generated with a FE of up to 6.8%. The three products generated from CO2 and SO32- suggested three possible reaction pathways.
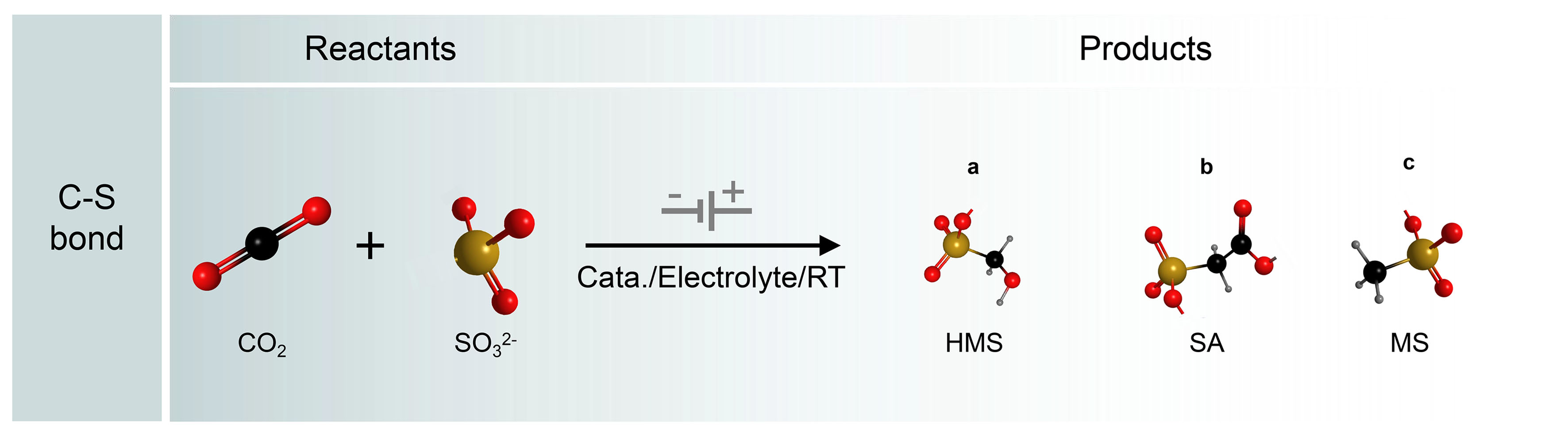
Figure 12. Electrocatalytic C−S coupling reactions and C−S products.
The relevant in-situ characterization experiments indicate that the C−S bond is formed by the attack of the SO32- ion in the solution on the *CHOH intermediate generated on the catalyst surface [Figure 13]. The authors speculate that the partially hydrogenated *CO (e.g., *CHO or CH2O) in the pathway of CO2 reduction to CH4 may be the main species coupled to SO32- to form HMS. SA and MS were preliminarily formed with SO32- nucleophilically attacking on C2 intermediates generated in the pathways of CO2 reduction to CH4 or acetic acid. Theoretical calculations reveal that among the intermediates (*COH, *CHO, and *CHOH) involved in C−S bond formation, coupling *CHOH with SO32- exhibited the lowest energy barrier (0.74 eV) over Cu (100), compared to *COH and SO32- coupling (1.37 eV). This indicates that *CHOH and SO32- coupling is the key step. Enhancing C−S coupling efficiency in future studies requires designing catalytically active sites to reduce energy barriers for *CHOH and SO32- coupling. Wu et al. underscored this study and remarked that it presents significant potential for synthesizing other high-value organics containing C−S bonds by adjusting CO2 reduction pathways or employing alternative catalysts and sulfur sources to minimize redundancy[146].
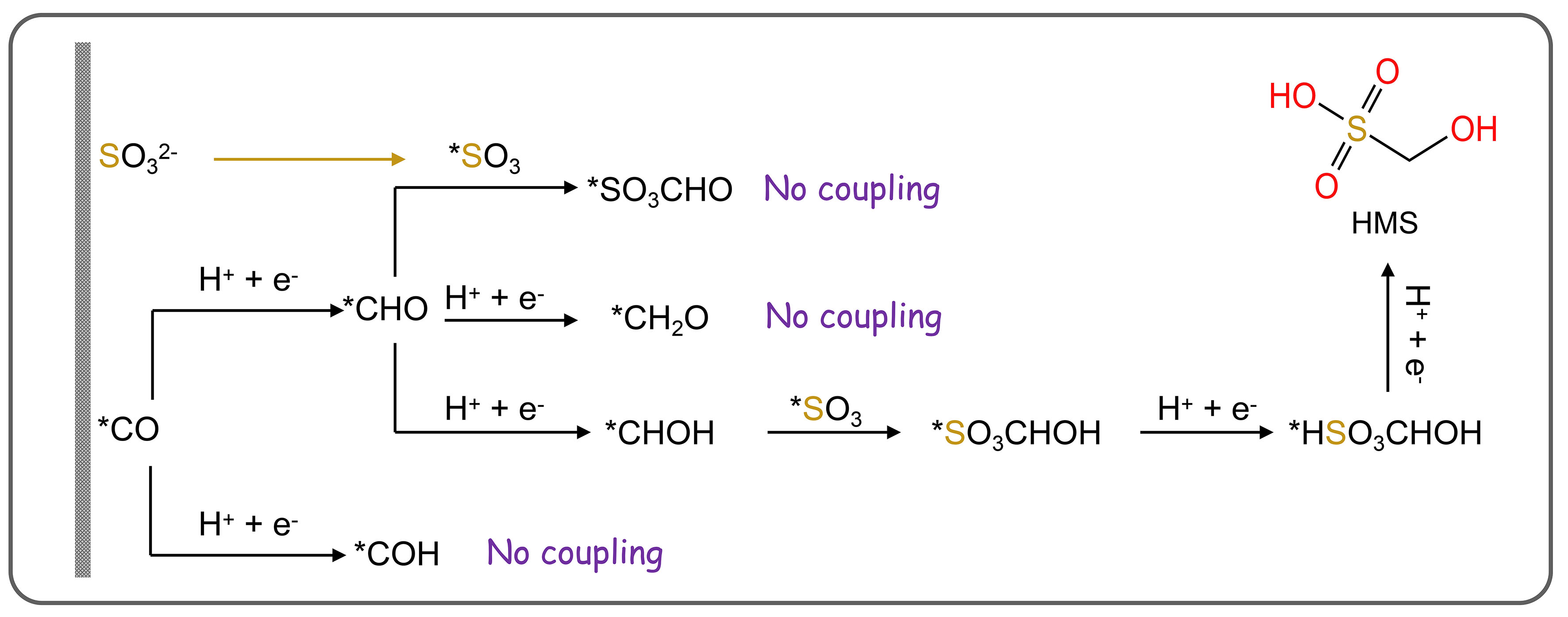
Figure 13. Reaction pathway from *CO to HMS. HMS: Hydroxymethanesulfonate.
EFFECT OF THE ELECTROLYTE FOR ELECTROCATALYTIC REACTION
The type of electrolyte can also significantly influence electrocatalytic performance. The primary reason is that different concentrations of anions and cations in the electrolyte can lead to variations in buffering capacity[147-149], pH value[150,151], and proton donors[152]. These factors often interact with each other, making it difficult to discuss the impact of any particular factor in detail. The following is a brief introduction to the effects of pH, cations, and anions in the electrolyte on the electrocatalytic CO2 reactions, using the CO2 reduction as a representative example.
As shown in Figure 14A, changes in proton donors (e.g., from H3O+ to H2O) or oxidants (e.g., from OH- to H2O) with pH can have an order-of-magnitude effect on the rate of electrochemical CO2 reduction steps. Electrochemical interfaces are, in general, highly charged, and these charges give rise to large interfacial electric fields at the electrode-electrolyte interface. These fields drive the transfer of electrons between the adsorbates involved in the reaction. The pH of the electrolyte also affects electrocatalytic activity through a solution-phase reaction with OH- without any involvement of the electrode[153]. When CO2 dissolves in an aqueous solution, different carbonaceous species such as CO2, H2CO3, HCO3-, and CO32- are formed, thus leading to debate about the true active species in the reaction process [Figure 14B][149,152]. Studies have shown that under alkaline conditions, over 50% of the energy is used to recover CO2 to CO32-. In contrast, acidic conditions inhibit the conversion of CO2 to CO32- and facilitate material separation. However, under strong acidic conditions, protons are more likely to capture electrons, thus favoring hydrogen evolution over CO2 reduction[154]. To date, the aqueous solutions used in most studies are weakly acidic or alkaline electrolytes containing cations and buffering/non-buffering anions [Figure 14C][149,155,156]. In these electrolytes, larger cations such as K+ and Cs+ could adsorb on the electrode and reduce hydrogen evolution by repelling H+ ions, whereas smaller cations such as Li+ do not. In addition, larger cations are hydrolyzed in the vicinity of the cathode, thereby lowering the pH and increasing the local CO2 concentration, increasing the product selectivity and reducing the formation of H2[155]. In addition to cations, anions also have a significant effect on the outcome of the electrocatalytic CO2 reaction. Non-buffering halides such as Cl- and Br- enhance the product selectivity by affecting surface charges and inhibiting proton adsorption, whereas F- negatively affects the product selectivity due to its weak adsorption capacity and high pH value. The use of anions with low buffering capacity inhibits the formation of H2 and thus improves the reaction efficiency[156].
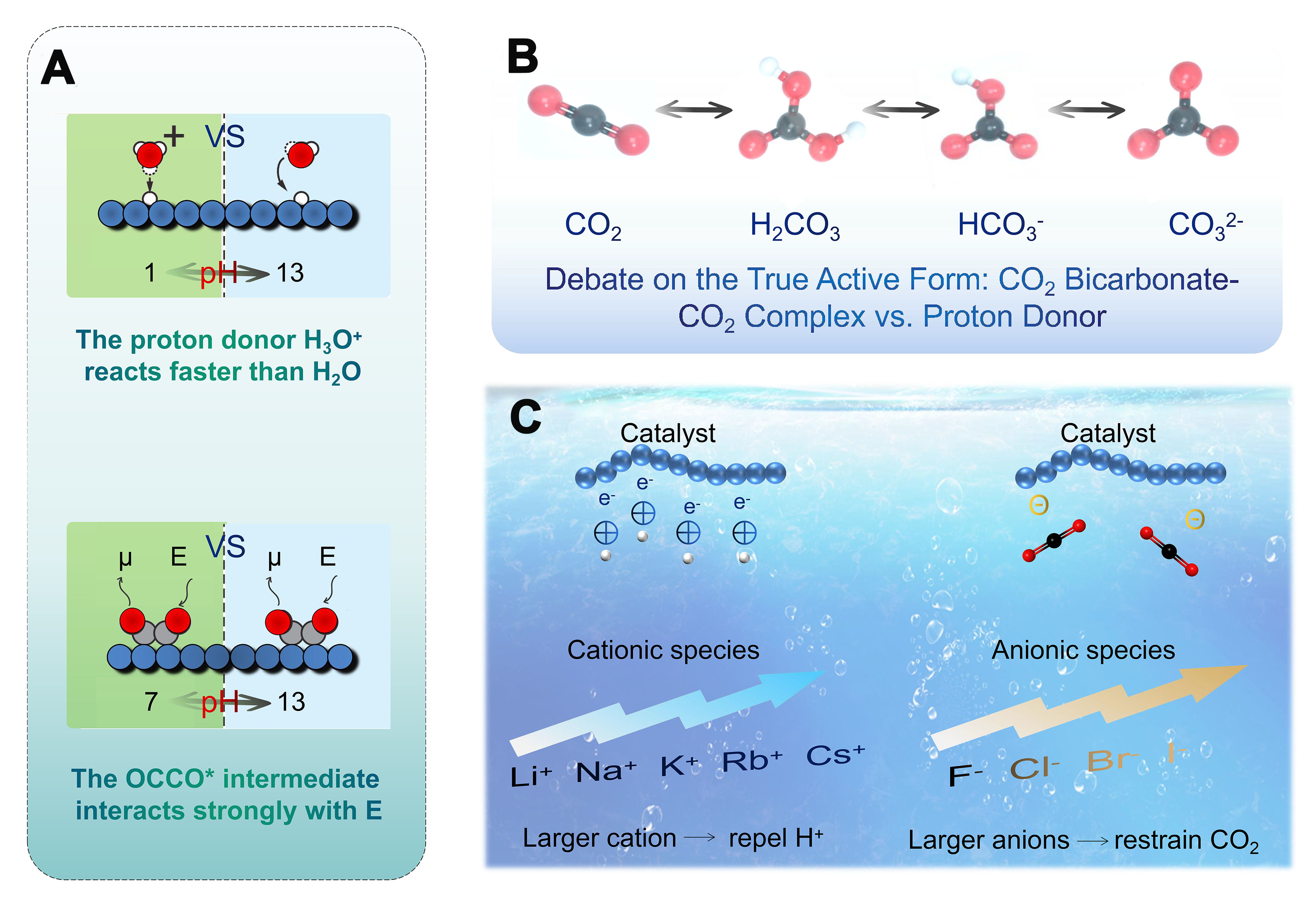
Figure 14. (A) The rate of electrocatalytic reactions can change with electrolyte pH primarily. Relative current densities are shown as bold numbers (Large electric fields E at high pH stabilize reactive intermediates with large dipole moments μ); (B) The discussion of CO2/H2CO3, H2CO3/HCO3-, and HCO3-/CO32- equilibria in aqueous solutions; (C) The cation and anion effects relating to the CO2 electroreduction.
DEVELOPMENT OF THE REACTOR IN THE ELECTROCATALYTIC REACTION
An in-depth study of the structural characteristics of different CO2 electrolyzers and facilitating their engineering are all key to large-scale applications[157]. Currently, the electrocatalytic performance is typically evaluated in H-cell electrolyzers, which employ an ion exchange membrane to separate anode and cathode compartments [Figure 15A][158-160]. However, the large distance between electrodes, membranes, and dilute electrolytes results in a high cell voltage and limited current density. To address these limitations, flow-cell
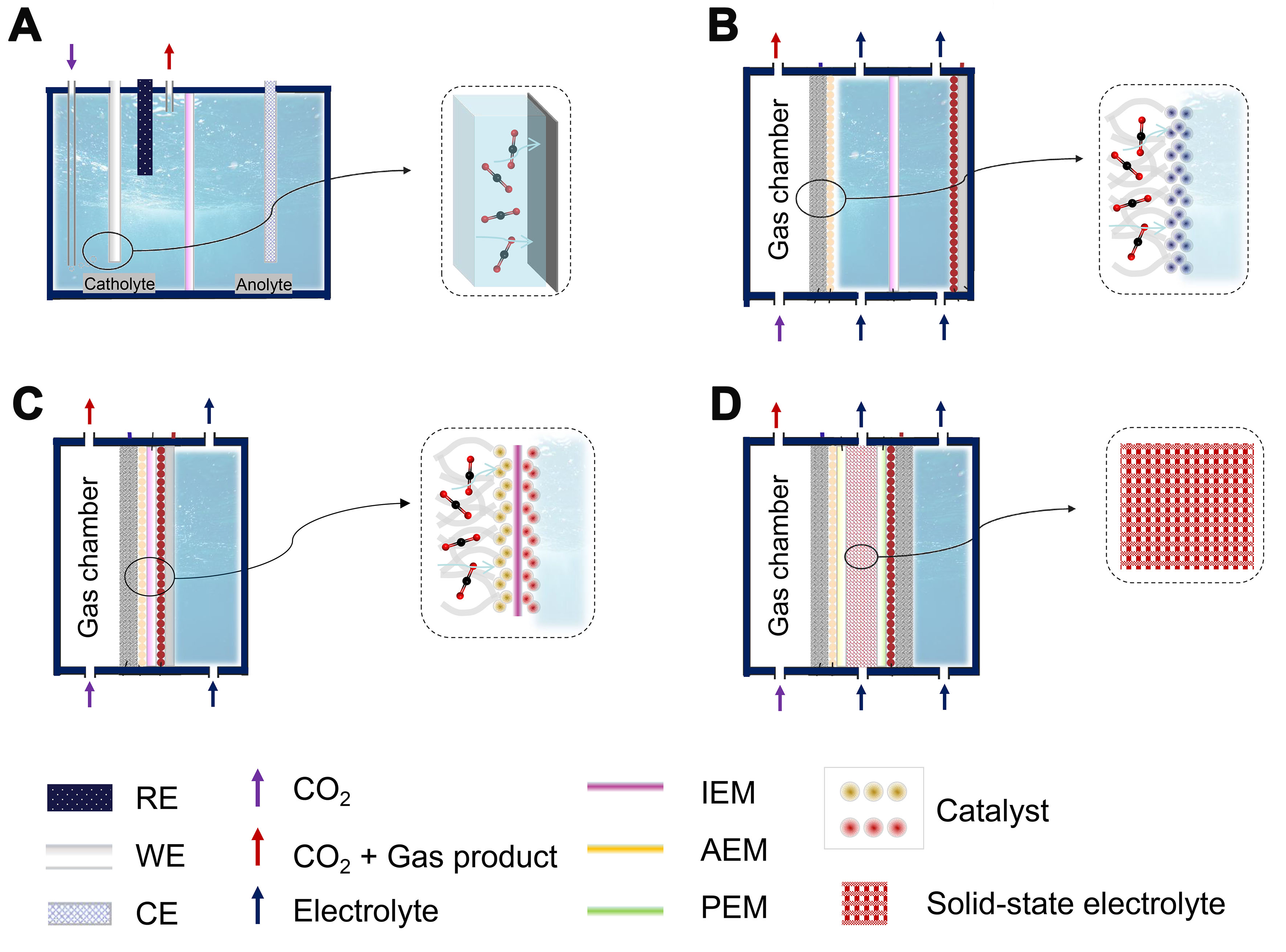
Figure 15. Different types of electrolyzers. (A) H-type electrolyzer; (B) GDE-type electrolyzer; (C) MEA-type electrolyzer; (D) Solid-state electrolyte type electrolyzer. GDE: Gas diffusion electrode; MEA: membrane electrode assembly; RE: reference electrode; WE: work electrode; CE: counter electrode; IEM: ion-exchange membranes; AEM: anion-exchange membrane; PEM: proton-exchange membrane.
According to the understanding of the above four types of electrolyzers and the review of related progress, we further summarize the advantages and disadvantages of each type [Figure 16], with the aim of accelerating the industrial application of CO2 electrocatalytic conversion technology.
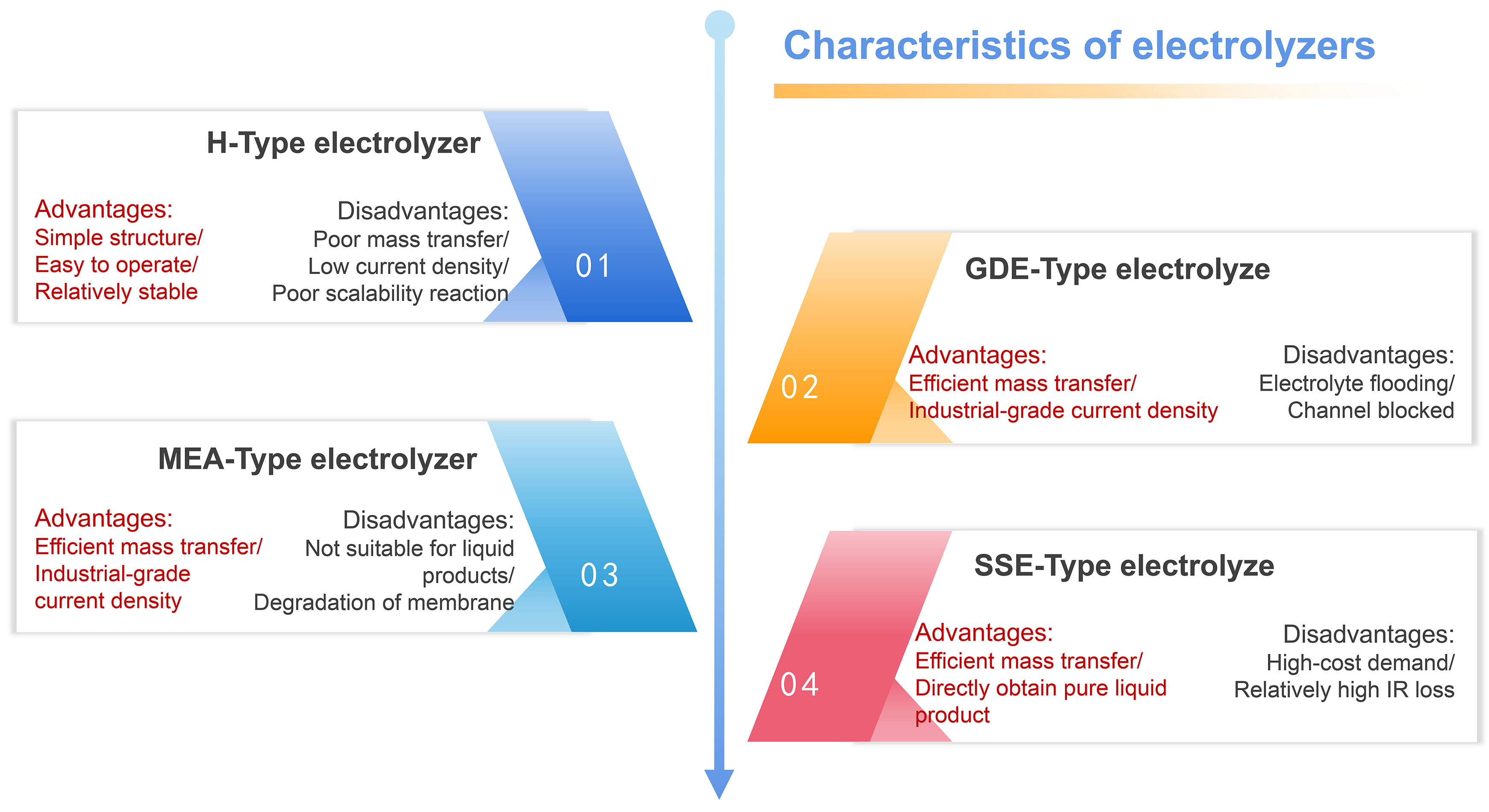
Figure 16. The advantages and disadvantages of electrolyzers.
CONCLUSIONS AND OUTLOOKS
In conclusion, the electrochemical conversion of CO2 into value-added chemicals is a promising way to address the severely increasing CO2 concentration in the atmosphere. New covalent bonds can be formed between CO2 and substrate molecules, including C−H, C−C, C−N, C−O and C−S bonds, thereby broadening reaction pathways and producing diversified products. In this review, we summarize the recent advances in the electrocatalysis-enabled CO2 cycle including the electrochemical C−X bond formation for high-value-added chemicals. We also discussed the fundamentals of electrocatalytic reactions on the initial activation of CO2 and focused on the research progress of electrochemical C−X (C−H, C−C, C−N, C−O, C−S) coupling reactions, proposing improvement strategies for the shortcomings of electrocatalytic CO2 coupled organic reactions and outlooks of their future research prospects. The following will give some views on (i) the strategies for the catalyst materials design and activity optimization and (ii) the future development of CO2 and small molecules to form five kinds of bonds.
(I) The strategies for the catalyst materials design and activity optimization
In order to optimize the design and activity of catalyst materials for the electrochemical conversion of CO2 into value-added chemicals, several advanced strategies can be employed. For example, nanostructural modification of catalysts can increase surface area and control morphology to improve catalytic activity and selectivity. Utilizing metallic and bimetallic catalysts as well as non-metallic options and heteroatom doping can modulate electronic properties and improve CO2 activation. Electronic structure engineering through defect and strain engineering can create active sites and improve performance. The use of composite and hybrid catalysts can improve electron transfer and stability. Specifically, the integration of multiple types of active sites into a single system is a simple and versatile strategy that allows for the independent optimization of intermediate adsorption and catalytic activity for each component reaction. By leveraging the synergistic effects between various active sites at the atomic scale, the coordination environment of atoms can be optimally adjusted. This approach provides active centers with varying charge distribution densities for the adsorption, activation, and conversion of different reactive molecules, and is becoming an important direction for the development of high-performance catalysts.
The following are some key factors in catalyst materials design:
(1) Active site design
Transition metal ligands: selecting appropriate transition metal ligands such as platinum, palladium, copper, etc., and their coordination environments can adjust the electrophilicity and coordination ability of active sites. Ancillary ligands: introducing different ancillary ligands can modulate the catalyst’s electronic structure and charge distribution, affecting the rate and selectivity of electron transfer.
(2) Surface modification
Synthesis methods: choosing suitable synthesis methods to control the crystal surface structure and surface chemical states of the catalyst can influence electron transfer capabilities and the specificity of chemical reactions. Surface engineering: enhancing catalyst surface site density and accessibility can improve reaction rates and selectivity.
(3) Control of electron transfer and energy barriers
Electron transfer pathways: designing catalysts to facilitate or hinder electron transfer can control the generation and dissociation of intermediates in reactions. Energy barrier tuning: adjusting the activation energy of reactions by modulating local electron densities on catalyst surfaces can enhance reaction rates and selectivity.
(4) Catalyst stability and regeneration
Enhancing stability: choosing stable catalyst materials and designing reaction conditions to minimize catalyst deactivation and corrosion. Regeneration strategies: developing effective catalyst regeneration methods to prolong catalyst lifetimes and reduce costs.
In addition, optimization of the electrolyte and design of an efficient catalyst-electrolyte interface can enhance reaction rates and reduce the energy barrier. Adjusting reaction conditions, including temperature, pressure, and applied potential, can optimize selectivity and efficiency. Improving stability and durability through corrosion-resistant materials and stable support structures ensures longer operational lifespans. Advanced characterization techniques and computational modeling provide deep insights into reaction mechanisms and guide the development of more effective catalysts. By implementing these strategies, researchers can significantly advance the field of electrocatalytic CO2 conversion to address environmental and economic challenges.
(II) The future development of CO2 and small molecules
(1) C−H bonds: currently, a variety of well-designed catalysts have been successfully used to convert CO2 into various C1 and C2 products, and the reaction efficiency has also been greatly improved, but for C3 and above products, the catalytic efficiency of the catalyst remains relatively low. The production of C2 products often requires coupling between C1 active species. If multiple C1 active species are required for coupling, the formation of C3, C4, and other products must be a huge challenge because it involves the complex transfer of multiple electrons and protons. If C1 and C2 products can be further activated on the catalyst, coupled themselves, or further combined with CO2, the primitive steps of the reaction will be greatly simplified, to achieve the synthesis of high-carbon products. For example, ethylene from the electrochemical conversion of CO2 can be activated by adsorption on a catalyst; then, there is the possibility of coupling between two or more molecules to obtain butane or high-carbon alkanes. The activated ethylene may also combine with one or two molecules of CO2 to produce propionic or succinic acid. Of course, the special structure of the catalyst and microenvironment of the system is very demanding, but for the synthesis of high-carbon products, this is an effective strategy.
(2) C−C bonds: the electrochemical carboxylation of CO2 with organic compounds is the main strategy to construct the C−C bond. Most of these organic compounds contain unsaturated bonds or highly polar covalent bonds, which help them to be activated at the cathode or easily bind to CO2 anion radicals. Such reactions usually employ ordinary graphite electrodes or bare metal sheet electrodes as cathodes, and the high selectivity of the product is obtained by optimizing the electrolyte solution. The larger the conjugated structure of the substrate or the stronger the polarity of the covalent bond, the higher the reactivity is. However, less research has been done on the carboxylation of substrates with non-conjugated structures and weakly polar covalent bonds. These electrochemically inert substrates are difficult to activate on the surface of ordinary electrodes and do not readily combine with the radical anion of CO2. Although some teams have recently realized the carboxylation of sp2 C−H bonds, the current efficiency is extremely low. For the carboxylation reaction of electrochemically inert substrates, it may be necessary to get rid of simple bare electrodes and rely on new electrocatalysts to activate the inert substrate and reduce its activation potential to achieve efficient binding with CO2. Based on current research progress, the application of electrocatalysts in electrochemical carboxylation reactions is still widely unexplored. However, it will be a powerful tool to explore the carboxylation of electrochemical inert substrates in the future and help to achieve highly selective carboxylation of common sp3 C−H bonds.
(3) C−N bonds: the reaction between CO2 and inorganic nitrogen sources to form urea by constructing C−N bonds is being widely explored. However, amides, another class of products that form new C−N bonds, have been less studied. The transition of C−N bond products from urea to amide by designing catalysts may become the focus of future research. The formation of the C−N bond does not have to be limited to the combination of CO2 and inorganic nitrogen sources, but CO2 can also be combined with organic nitrogen sources to achieve the functional group of organic compounds. Furthermore, enhancing the efficiency, cost-effectiveness, and environmental sustainability of catalytic systems for large-scale electrosynthesis of organic nitrogen compounds via C−N coupling reactions under mild conditions requires further investigation.
(4) C−O bonds: the reaction of alcohols or epoxides with CO2 to form carbonates by constructing C−O bonds has been extensively studied. By adjusting the electrode and electrolyte, the target product can be obtained with high selectivity. Cyclic carbonates are in high demand as monomers for polymers. The strategy of obtaining cyclic carbonates directly from CO2 is extremely attractive, but there are few related studies. In principle, CO2 can be converted into ethylene oxide by pairwise reaction of anode and cathode. By further combining ethylene oxide with CO2, cyclic carbonates can be obtained. In the future, this will be an attractive green synthetic strategy.
(5) C−S bonds: at present, the electrolyte system of this reaction was alkaline, which will cause low CO2 utilization efficiency. Acid electrolyte system can be explored to improve the utilization efficiency of CO2. On the other hand, it is necessary to design efficient catalysts and electrolyzers to improve the FEs of C–S products and understand the mechanism of the reaction. Finally, the types of S sources can be explored, which may result in more diverse products.
Meanwhile, to advance the field, great achievements have been made in understanding the reaction pathways of electrocatalytic CO2 conversion using advanced in situ and operational techniques and theoretical calculations. To further explain the catalytic mechanism, analyzing the reaction pathways, surface charges, and free energies on the catalyst surface by constructing emerging operational computational models can be beneficial for us to better understand the energy and thermodynamics of non-uniform electrocatalytic reactions, such as the potential dependence of the activation energy, the interactions between intermediate coverings, and the adsorption-adsorption interactions.
More importantly, the following factors should be considered for future large-scale application of electrocatalytic production: (i) the decreasing price of renewable electrical energy; (ii) developing low-cost and long-term stable catalysts; (iii) avoiding use of harmful organic solvent and additives; (iv) preparing efficient electrolyzers and durable membranes; and (v) solving the problem of product separation and purification. In this regard, direct-current electrolyzer units have shown great advantages in realizing industrial-scale currents and hold promise for large-scale production of valuable fuels and high-value-added chemicals.
DECLARATIONS
Authors’ contributions
Manuscript preparation: Jia S, Dong M
Manuscript correction: Zhu Q, Kang X, Wu H, Han B
All authors contributed to the general discussion.
Availability of data and materials
Not applicable.
Financial support and sponsorship
The work was supported by the National Key Research and Development Program of China (2023YFA1507440, 2020YFA0710201), the National Natural Science Foundation of China (22279146, 21890761, 22121002), the China Postdoctoral Science Foundation (2023M731096), and the Fundamental Research Funds for the Central Universities.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. He M, Sun Y, Han B. Green carbon science: scientific basis for integrating carbon resource processing, utilization, and recycling. Angew Chem Int Ed Engl. 2013;52:9620-33.
2. Wang Y, Wang C, Li M, Yu Y, Zhang B. Nitrate electroreduction: mechanism insight, in situ characterization, performance evaluation, and challenges. Chem Soc Rev. 2021;50:6720-33.
3. Huang Y, Wang Y, Wu Y, Yu Y, Zhang B. Electrocatalytic construction of the C-N bond from the derivates of CO2 and N2. Sci China Chem. 2022;65:204-6.
4. Pang Y, Li J, Wang Z, et al. Efficient electrocatalytic conversion of carbon monoxide to propanol using fragmented copper. Nat Catal. 2019;2:251-8.
5. Ran CK, Xiao HZ, Liao LL, Ju T, Zhang W, Yu DG. Progress and challenges in dicarboxylation with CO2. NSO. 2023;2:20220024.
6. Zhou Z, Pei Z, Wei L, Zhao S, Jian X, Chen Y. Electrocatalytic hydrogen evolution under neutral pH conditions: current understandings, recent advances, and future prospects. Energy Environ Sci. 2020;13:3185-206.
7. Guo N, Xue H, Bao A, et al. Achieving superior electrocatalytic performance by surface copper vacancy defects during electrochemical etching process. Angew Chem Int Ed Engl. 2020;59:13778-84.
8. Zhou Y, Che F, Liu M, et al. Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat Chem. 2018;10:974-80.
9. Yang D, Zhu Q, Han B. Electroreduction of CO2 in ionic liquid-based electrolytes. Innovation. 2020;1:100016.
10. Wang Q, Wang Y, Liu M, Chu G, Qiu Y. Recent advances in photochemical/electrochemical carboxylation of olefins with CO2. Chin J Chem. 2024;42:2249-66.
11. Jin H, Yu H, Li H, et al. MXene analogue: a 2D nitridene solid solution for high-rate hydrogen production. Angew Chem Int Ed Engl. 2022;61:e202203850.
12. Chang S, Bu J, Li J, et al. Highly efficient electrocatalytic deuteration of acetylene to deuterated ethylene using deuterium oxide. Chin Chem Lett. 2023;34:107765.
13. Zeng W, Qiu Y. Electrochemical conversion of organic compounds and inorganic small molecules. Sci China Chem. 2024;67:3223-46.
14. Liu X, Zhang K, Tao L, Lu X, Zhang W. Recent advances in electrochemical carboxylation reactions using carbon dioxide. Green Chem Eng. 2022;3:125-37.
15. Zhao Z, Liu Y, Wang S, et al. Site-selective electrochemical C-H carboxylation of arenes with CO2. Angew Chem Int Ed Engl. 2023;62:e202214710.
16. Zhang W, Liao LL, Li L, et al. Electroreductive dicarboxylation of unactivated skipped dienes with CO2. Angew Chem Int Ed Engl. 2023;62:e202301892.
17. Wang Y, Tang S, Yang G, Wang S, Ma D, Qiu Y. Electrocarboxylation of aryl epoxides with CO2 for the facile and selective synthesis of β-hydroxy acids. Angew Chem Int Ed Engl. 2022;61:e202207746.
18. Zhou H, Ren Y, Li Z, et al. Electrocatalytic upcycling of polyethylene terephthalate to commodity chemicals and H2 fuel. Nat Commun. 2021;12:4679.
19. Wang T, Tao L, Zhu X, et al. Combined anodic and cathodic hydrogen production from aldehyde oxidation and hydrogen evolution reaction. Nat Catal. 2022;5:66-73.
20. Zhang B, Jiang Y, Gao M, Ma T, Sun W, Pan H. Recent progress on hybrid electrocatalysts for efficient electrochemical CO2 reduction. Nano Energy. 2021;80:105504.
21. Singh K, Sohal HS, Singh B. Synthesis of α-hydroxycarboxylic acids from various aldehydes and ketones by direct electrocarboxylation: a facile, efficient and atom economy protocol. Asian J Chem. 2021;33:839-45.
22. Krzywda PM, Paradelo Rodríguez A, Benes NE, Mei BT, Mul G. Carbon-nitrogen bond formation on Cu electrodes during CO2 reduction in NO3- solution. Appl Catal B Environ. 2022;316:121512.
23. Li JY, Song QW, Zhang K, Liu P. Catalytic conversion of carbon dioxide through C-N bond formation. Molecules. 2019;24:182.
24. Li R, Xiang K, Liu Z, Peng Z, Zou Y, Wang S. Recent advances in upgrading of low-cost oxidants to value-added products by electrocatalytic reduction reaction. Adv Funct Mater. 2022;32:2208212.
25. You B, Sun Y. Innovative strategies for electrocatalytic water splitting. Acc Chem Res. 2018;51:1571-80.
26. Seh ZW, Kibsgaard J, Dickens CF, Chorkendorff I, Nørskov JK, Jaramillo TF. Combining theory and experiment in electrocatalysis: insights into materials design. Science. 2017;355:eaad4998.
27. Zhang H, Chang X, Chen JG, et al. Computational and experimental demonstrations of one-pot tandem catalysis for electrochemical carbon dioxide reduction to methane. Nat Commun. 2019;10:3340.
28. Wang X, de Araújo JF, Ju W, et al. Mechanistic reaction pathways of enhanced ethylene yields during electroreduction of CO2-CO co-feeds on Cu and Cu-tandem electrocatalysts. Nat Nanotechnol. 2019;14:1063-70.
29. Tang C, Zheng Y, Jaroniec M, Qiao SZ. Electrocatalytic refinery for sustainable production of fuels and chemicals. Angew Chem Int Ed Engl. 2021;60:19572-90.
30. Peng C, Luo G, Zhang J, et al. Double sulfur vacancies by lithium tuning enhance CO2 electroreduction to n-propanol. Nat Commun. 2021;12:1580.
31. Muchez L, De Vos DE, Kim M. Sacrificial anode-free electrosynthesis of α-hydroxy acids via electrocatalytic coupling of carbon dioxide to aromatic alcohols. ACS Sustainable Chem Eng. 2019;7:15860-4.
32. Li Z, Qu Y, Wang J, et al. Highly selective conversion of carbon dioxide to aromatics over tandem catalysts. Joule. 2019;3:570-83.
33. Lum Y, Ager JW. Sequential catalysis controls selectivity in electrochemical CO2 reduction on Cu. Energy Environ Sci. 2018;11:2935-44.
34. Leow WR, Lum Y, Ozden A, et al. Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density. Science. 2020;368:1228-33.
35. Wu Y, Jiang Z, Lin Z, Liang Y, Wang H. Direct electrosynthesis of methylamine from carbon dioxide and nitrate. Nat Sustain. 2021;4:725-30.
36. Deng T, Jia S, Han S, et al. Electrochemical CO2 reduction to C2+ products over Cu/Zn intermetallic catalysts synthesized by electrodeposition. Front Energy. 2024;18:80-8.
37. De Luna P, Quintero-Bermudez R, Dinh CT, et al. Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nat Catal. 2018;1:103-10.
38. Zhu Q, Ma J, Kang X, et al. Efficient reduction of CO2 into formic acid on a lead or tin electrode using an ionic liquid catholyte mixture. Angew Chem Int Ed Engl. 2016;55:9012-6.
39. Zhang S, Fan Q, Xia R, Meyer TJ. CO2 reduction: from homogeneous to heterogeneous electrocatalysis. Acc Chem Res. 2020;53:255-64.
40. Kim D, Xie C, Becknell N, et al. Electrochemical activation of CO2 through atomic ordering transformations of AuCu nanoparticles. J Am Chem Soc. 2017;139:8329-36.
41. Gao S, Lin Y, Jiao X, et al. Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel. Nature. 2016;529:68-71.
42. Gao D, Zhou H, Wang J, et al. Size-dependent electrocatalytic reduction of CO2 over Pd nanoparticles. J Am Chem Soc. 2015;137:4288-91.
43. Lu Q, Rosen J, Zhou Y, et al. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat Commun. 2014;5:3242.
44. Medina-Ramos J, DiMeglio JL, Rosenthal J. Efficient reduction of CO2 to CO with high current density using in situ or ex situ prepared Bi-based materials. J Am Chem Soc. 2014;136:8361-7.
45. Ma W, Xie S, Liu T, et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C-C coupling over fluorine-modified copper. Nat Catal. 2020;3:478-87.
46. Zhang S, Kang P, Meyer TJ. Nanostructured tin catalysts for selective electrochemical reduction of carbon dioxide to formate. J Am Chem Soc. 2014;136:1734-7.
47. Wu Y, Jiang Z, Lu X, Liang Y, Wang H. Domino electroreduction of CO2 to methanol on a molecular catalyst. Nature. 2019;575:639-42.
48. Jia S, Zhu Q, Wu H, et al. Preparation of trimetallic electrocatalysts by one-step co-electrodeposition and efficient CO2 reduction to ethylene. Chem Sci. 2022;13:7509-15.
49. Song Y, Chen W, Zhao C, Li S, Wei W, Sun Y. Metal-free nitrogen-doped mesoporous carbon for electroreduction of CO2 to ethanol. Angew Chem Int Ed Engl. 2017;56:10840-4.
50. Jia S, Zhu Q, Chu M, et al. Hierarchical metal-polymer hybrids for enhanced CO2 electroreduction. Angew Chem Int Ed Engl. 2021;60:10977-82.
51. Nam DH, De Luna P, Rosas-Hernández A, et al. Molecular enhancement of heterogeneous CO2 reduction. Nat Mater. 2020;19:266-76.
52. Li F, Thevenon A, Rosas-Hernández A, et al. Molecular tuning of CO2-to-ethylene conversion. Nature. 2020;577:509-13.
53. Ren S, Joulié D, Salvatore D, et al. Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell. Science. 2019;365:367-9.
54. Nørskov JK, Bligaard T, Logadottir A, et al. Trends in the exchange current for hydrogen evolution. J Electrochem Soc. 2005;152:J23.
55. Shi R, Wang Z, Zhao Y, et al. Room-temperature electrochemical acetylene reduction to ethylene with high conversion and selectivity. Nat Catal. 2021;4:565-74.
56. Zhang Z, Wen G, Luo D, et al. “Two ships in a bottle” design for Zn-Ag-O catalyst enabling selective and long-lasting CO2 electroreduction. J Am Chem Soc. 2021;143:6855-64.
57. Ren W, Tan X, Yang W, et al. Isolated diatomic Ni-Fe metal-nitrogen sites for synergistic electroreduction of CO2. Angew Chem Int Ed Engl. 2019;58:6972-6.
58. Hu C, Gao Y, Zhao L, Dai L. Carbon-based metal-free electrocatalysts: recent progress and forward looking. Chem Catal. 2022;2:2150-6.
59. Kondo T, Casolo S, Suzuki T, et al. Atomic-scale characterization of nitrogen-doped graphite: effects of dopant nitrogen on the local electronic structure of the surrounding carbon atoms. Phys Rev B. 2012;86:035436.
60. Guo D, Shibuya R, Akiba C, Saji S, Kondo T, Nakamura J. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science. 2016;351:361-5.
61. Bagger A, Ju W, Varela AS, Strasser P, Rossmeisl J. Electrochemical CO2 reduction: a classification problem. Chemphyschem. 2017;18:3266-73.
62. Katayama Y, Nattino F, Giordano L, et al. An in situ surface-enhanced infrared absorption spectroscopy study of electrochemical CO2 reduction: selectivity dependence on surface C-bound and O-bound reaction intermediates. J Phys Chem C. 2019;123:5951-63.
63. Lin R, Ma X, Cheong WC, et al. PdAg bimetallic electrocatalyst for highly selective reduction of CO2 with low COOH* formation energy and facile CO desorption. Nano Res. 2019;12:2866-71.
64. Deng T, Jia S, Chen C, et al. Polymer modification strategy to modulate reaction microenvironment for enhanced CO2 electroreduction to ethylene. Angew Chem Int Ed Engl. 2024;63:e202313796.
65. Chen X, Jia S, Chen C, et al. Highly stable layered coordination polymer electrocatalyst toward efficient CO2-to-CH4 conversion. Adv Mater. 2024;36:e2310273.
66. de Arquer FPG, Dinh CT, Ozden A, et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm-2. Science. 2020;367:661-6.
67. Li W, Yin Z, Gao Z, et al. Bifunctional ionomers for efficient co-electrolysis of CO2 and pure water towards ethylene production at industrial-scale current densities. Nat Energy. 2022;7:835-43.
68. Endrődi B, Kecsenovity E, Samu A, et al. High carbonate ion conductance of a robust PiperION membrane allows industrial current density and conversion in a zero-gap carbon dioxide electrolyzer cell. Energy Environ Sci. 2020;13:4098-105.
69. Zhang Y, Zhang XY, Sun WY. In situ carbon-encapsulated copper-doped cerium oxide derived from MOFs for boosting CO2-to-CH4 electro-conversion. ACS Catal. 2023;13:1545-53.
70. Jiang Y, Mao K, Li J, et al. Pushing the performance limit of Cu/CeO2 catalyst in CO2 electroreduction: a cluster model study for loading single atoms. ACS Nano. 2023;17:2620-8.
71. Li H, Cao S, Sun H, et al. CuNCN derived Cu-based/CxNy catalysts for highly selective CO2 electroreduction to hydrocarbons. Appl Catal B Environ. 2023;320:121948.
72. Zou H, Zhao G, Dai H, et al. Electronic perturbation of copper single-atom CO2 reduction catalysts in a molecular way. Angew Chem Int Ed Engl. 2023;62:e202217220.
73. Shi G, Xie Y, Du L, et al. Constructing Cu-C bonds in a graphdiyne-regulated cu single-atom electrocatalyst for CO2 reduction to CH4. Angew Chem Int Ed Engl. 2022;61:e202203569.
74. Wang Z, Zhou Y, Xia C, Guo W, You B, Xia BY. Efficient electroconversion of carbon dioxide to formate by a reconstructed amino-functionalized indium-organic framework electrocatalyst. Angew Chem Int Ed Engl. 2021;60:19107-12.
75. Cui K, Zhang Z, Wang C, Lyu P, Tang X, Xu Y. Modulating the D-π-A interactions in metal-covalent organic frameworks for efficient electroreduction of CO2 into formate. Angew Chem Int Ed Engl. 2024;63:e202407298.
76. Zhu ZH, Zhao BH, Hou SL, et al. A facile strategy for constructing a carbon-particle-modified metal-organic framework for enhancing the efficiency of CO2 electroreduction into formate. Angew Chem Int Ed Engl. 2021;60:23394-402.
77. Luo W, Xie W, Mutschler R, et al. Selective and stable electroreduction of CO2 to CO at the copper/indium interface. ACS Catal. 2018;8:6571-81.
78. Rasul S, Anjum DH, Jedidi A, Minenkov Y, Cavallo L, Takanabe K. A highly selective copper-indium bimetallic electrocatalyst for the electrochemical reduction of aqueous CO2 to CO. Angew Chem Int Ed Engl. 2015;54:2146-50.
79. Yan C, Li H, Ye Y, et al. Coordinatively unsaturated nickel-nitrogen sites towards selective and high-rate CO2 electroreduction. Energy Environ Sci. 2018;11:1204-10.
80. Jia S, Zhu Q, Han S, et al. Ultra-fast synthesis of three-dimensional porous Cu/Zn heterostructures for enhanced carbon dioxide electroreduction. Chem Sci. 2023;14:11474-80.
81. Liu C, Zhang XD, Huang JM, Guan MX, Xu M, Gu ZY. In situ reconstruction of Cu-N coordinated MOFs to generate dispersive Cu/Cu2O nanoclusters for selective electroreduction of CO2 to C2H4. ACS Catal. 2022;12:15230-40.
82. Zhang XD, Liu T, Liu C, et al. Asymmetric low-frequency pulsed strategy enables ultralong CO2 reduction stability and controllable product selectivity. J Am Chem Soc. 2023;145:2195-206.
83. He Q, Li H, Hu Z, Lei L, Wang D, Li TT. Highly selective CO2 electroreduction to C2H4 using a dual-sites Cu(II) porphyrin framework coupled with Cu2O nanoparticles via a synergetic-tandem strategy. Angew Chem Int Ed Engl. 2024;63:e202407090.
84. Zhang Y, Li P, Zhao C, et al. Multicarbons generation factory: CuO/Ni single atoms tandem catalyst for boosting the productivity of CO2 electrocatalysis. Sci Bull. 2022;67:1679-87.
85. Xu H, Rebollar D, He H, et al. Highly selective electrocatalytic CO2 reduction to ethanol by metallic clusters dynamically formed from atomically dispersed copper. Nat Energy. 2020;5:623-32.
86. Gu Z, Shen H, Chen Z, et al. Efficient electrocatalytic CO2 reduction to C2+ alcohols at defect-site-rich Cu surface. Joule. 2021;5:429-40.
87. Guo C, Guo Y, Shi Y, et al. Electrocatalytic reduction of CO2 to ethanol at close to theoretical potential via engineering abundant electron-donating Cuδ+ species. Angew Chem Int Ed Engl. 2022;61:e202205909.
88. Qiu XF, Huang JR, Yu C, et al. A stable and conductive covalent organic framework with isolated active sites for highly selective electroreduction of carbon dioxide to acetate. Angew Chem Int Ed Engl. 2022;61:e202206470.
89. Liu Y, Chen S, Quan X, Yu H. Efficient electrochemical reduction of carbon dioxide to acetate on nitrogen-doped nanodiamond. J Am Chem Soc. 2015;137:11631-6.
90. Sun X, Zhu Q, Kang X, et al. Design of a Cu(i)/C-doped boron nitride electrocatalyst for efficient conversion of CO2 into acetic acid. Green Chem. 2017;19:2086-91.
91. Zhang R, Zhang J, Wang S, et al. Synthesis of n-Propanol from CO2 electroreduction on bicontinuous Cu2O/Cu nanodomains. Angew Chem Int Ed Engl. 2024;63:e202405733.
92. Alkayal A, Tabas V, Montanaro S, Wright IA, Malkov AV, Buckley BR. Harnessing applied potential: selective
93. Sheta AM, Alkayal A, Mashaly MA, et al. Selective electrosynthetic hydrocarboxylation of α,β-unsaturated esters with carbon dioxide**. Angew Chem Int Ed Engl. 2021;60:21832-7.
94. Sheta AM, Mashaly MA, Said SB, Elmorsy SS, Malkov AV, Buckley BR. Selective α,δ-hydrocarboxylation of conjugated dienes utilizing CO2 and electrosynthesis. Chem Sci. 2020;11:9109-14.
95. Liao LL, Wang ZH, Cao KG, et al. Electrochemical ring-opening dicarboxylation of strained carbon-carbon single bonds with CO2: facile synthesis of diacids and derivatization into polyesters. J Am Chem Soc. 2022;144:2062-8.
96. Sun GQ, Zhang W, Liao LL, et al. Nickel-catalyzed electrochemical carboxylation of unactivated aryl and alkyl halides with CO2. Nat Commun. 2021;12:7086.
97. Wang Y, Zhao Z, Pan D, et al. Metal-free electrochemical carboxylation of organic halides in the presence of catalytic amounts of an organomediator. Angew Chem Int Ed Engl. 2022;61:e202210201.
98. Sun GQ, Yu P, Zhang W, et al. Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations. Nature. 2023;615:67-72.
99. Yuan G, Jiang H, Lin C, Liao S. Efficient electrochemical synthesis of 2-arylsuccinic acids from CO2 and aryl-substituted alkenes with nickel as the cathode. Electrochimica Acta. 2008;53:2170-6.
100. Li C, Yuan G, Ji X, Wang X, Ye J, Jiang H. Highly regioselective electrochemical synthesis of dioic acids from dienes and carbon dioxide. Electrochim Acta. 2011;56:1529-34.
101. Chen BL, Tu ZY, Zhu HW, Sun WW, Wang H, Lu JX. CO2 as a C1-organic building block: Enantioselective electrocarboxylation of aromatic ketones with CO2catalyzed by cinchona alkaloids under mild conditions. Electrochimica Acta. 2014;116:475-83.
102. Feng Q, Huang K, Liu S, Yu J, Liu F. Electrocatalytic carboxylation of aromatic ketones with carbon dioxide in ionic liquid 1-butyl-3-methylimidazoliumtetrafluoborate to α-hydroxy-carboxylic acid methyl ester. Electrochim Acta. 2011;56:5137-41.
103. Yang H, Wu L, Wang H, Lu J. Cathode made of compacted silver nanoparticles for electrocatalytic carboxylation of 1-phenethyl bromide with CO2. Chin J Catal. 2016;37:994-8.
104. Li C, Song X, Tao L, et al. Electrogenerated-bases promoted electrochemical synthesis of N-bromoamino acids from imines and carbon dioxide. Tetrahedron. 2014;70:1855-60.
105. Xie SL, Gao XT, Wu HH, Zhou F, Zhou J. Direct electrochemical defluorinative carboxylation of gem-difluoroalkenes with carbon dioxide. Org Lett. 2020;22:8424-9.
106. Ang NWJ, Oliveira JCA, Ackermann L. Electroreductive cobalt-catalyzed carboxylation: cross-electrophile electrocoupling with atmospheric CO2. Angew Chem Int Ed Engl. 2020;59:12842-7.
107. Yang D, Zhu M, Schiffer ZJ, et al. Direct electrochemical carboxylation of benzylic C−N bonds with carbon dioxide. ACS Catal. 2019;9:4699-705.
108. Li C, Yuan G, Jiang H. Electrocarboxylation of alkynes with carbon dioxide in the presence of metal salt catalysts. Chin J Chem. 2010;28:1685-9.
109. Kong Y, Wei W, Xu L, Chen C. Electrochemical synthesis of organonitrogen compounds from N-integrated CO2 reduction reaction. Acta Phys Chim Sin. 2024;40:2307049.
110. Yuan M, Chen J, Bai Y, et al. Unveiling electrochemical urea synthesis by co-activation of CO2 and N2 with mott-schottky heterostructure catalysts. Angew Chem Int Ed Engl. 2021;60:10910-8.
111. Jouny M, Lv JJ, Cheng T, et al. Formation of carbon-nitrogen bonds in carbon monoxide electrolysis. Nat Chem. 2019;11:846-51.
112. Li J, Kornienko N. Electrochemically driven C−N bond formation from CO2 and ammonia at the triple-phase boundary. Chem Sci. 2022;13:3957-64.
113. Yu Y, Lv Z, Liu Z, et al. Activation of Ga liquid catalyst with continuously exposed active sites for electrocatalytic C-N coupling. Angew Chem Int Ed Engl. 2024;63:e202402236.
114. Yuan M, Chen J, Zhang H, et al. Host-guest molecular interaction promoted urea electrosynthesis over a precisely designed conductive metal-organic framework. Energy Environ Sci. 2022;15:2084-95.
115. Yuan M, Zhang H, Xu Y, et al. Artificial frustrated Lewis pairs facilitating the electrochemical N2 and CO2 conversion to urea. Chem Catal. 2022;2:309-20.
116. Liu S, Yin S, Wang Z, et al. AuCu nanofibers for electrosynthesis of urea from carbon dioxide and nitrite. Cell Rep Phys Sci. 2022;3:100869.
117. Meng N, Huang Y, Liu Y, Yu Y, Zhang B. Electrosynthesis of urea from nitrite and CO2 over oxygen vacancy-rich ZnO porous nanosheets. Cell Rep Phys Sci. 2021;2:100378.
118. Saravanakumar D, Song J, Lee S, Hur NH, Shin W. Electrocatalytic conversion of carbon dioxide and nitrate ions to urea by a titania-nafion composite electrode. ChemSusChem. 2017;10:3999-4003.
119. Chen C, Zhu X, Wen X, et al. Coupling N2 and CO2 in H2O to synthesize urea under ambient conditions. Nat Chem. 2020;12:717-24.
120. Shibata M, Yoshida K, Furuya N. Electrochemical synthesis of urea on reduction of carbon dioxide with nitrate and nitrite ions using Cu-loaded gas-diffusion electrode. J Electroanal Chem. 1995;387:143-5.
121. Shibata M, Furuya N. Electrochemical synthesis of urea at gas-diffusion electrodes: Part VI. Simultaneous reduction of carbon dioxide and nitrite ions with various metallophthalocyanine catalysts. J Electroanal Chem. 2001;507:177-84.
122. Huang Y, Yang R, Wang C, et al. Direct electrosynthesis of urea from carbon dioxide and nitric oxide. ACS Energy Lett. 2022;7:284-91.
123. Lv Z, Zhou S, Zhao L, et al. Coactivation of multiphase reactants for the electrosynthesis of urea. Adv Energy Mater. 2023;13:2300946.
124. Yuan M, Chen J, Bai Y, et al. Electrochemical C-N coupling with perovskite hybrids toward efficient urea synthesis. Chem Sci. 2021;12:6048-58.
125. Lv C, Zhong L, Liu H, et al. Selective electrocatalytic synthesis of urea with nitrate and carbon dioxide. Nat Sustain. 2021;4:868-76.
126. Xu M, Wu F, Zhang Y, et al. Kinetically matched C-N coupling toward efficient urea electrosynthesis enabled on copper single-atom alloy. Nat Commun. 2023;14:6994.
127. Guo C, Zhou W, Lan X, et al. Electrochemical upgrading of formic acid to formamide via coupling nitrite co-reduction. J Am Chem Soc. 2022;144:16006-11.
128. Shao J, Meng N, Wang Y, et al. Scalable electrosynthesis of formamide through C-N coupling at the industrially relevant current density of 120 mA cm-2. Angew Chem Int Ed Engl. 2022;61:e202213009.
129. Fang Y, Liu X, Liu Z, et al. Synthesis of amino acids by electrocatalytic reduction of CO2 on chiral Cu surfaces. Chem. 2023;9:460-71.
130. Han L, Wu Y, Fang K, et al. The splanchnic mesenchyme is the tissue of origin for pancreatic fibroblasts during homeostasis and tumorigenesis. Nat Commun. 2023;14:1.
131. Weng LC, Bell AT, Weber AZ. Towards membrane-electrode assembly systems for CO2 reduction: a modeling study. Energy Environ Sci. 2019;12:1950-68.
132. Garg S, Li M, Weber AZ, et al. Advances and challenges in electrochemical CO2 reduction processes: an engineering and design perspective looking beyond new catalyst materials. J Mater Chem A. 2020;8:1511-44.
133. Mo Y, Lu Z, Rughoobur G, et al. Microfluidic electrochemistry for single-electron transfer redox-neutral reactions. Science. 2020;368:1352-7.
134. Vara BA, Struble TJ, Wang W, Dobish MC, Johnston JN. Enantioselective small molecule synthesis by carbon dioxide fixation using a dual Brønsted acid/base organocatalyst. J Am Chem Soc. 2015;137:7302-5.
135. Yousefi R, Struble TJ, Payne JL, Vishe M, Schley ND, Johnston JN. Catalytic, enantioselective synthesis of cyclic carbamates from dialkyl amines by CO2-capture: discovery, development, and mechanism. J Am Chem Soc. 2019;141:618-25.
136. Bansode A, Urakawa A. Continuous DMC synthesis from CO2 and methanol over a CeO2 catalyst in a fixed bed reactor in the presence of a dehydrating agent. ACS Catal. 2014;4:3877-80.
137. Selva M, Perosa A, Rodríguez-padrón D, Luque R. Applications of dimethyl carbonate for the chemical upgrading of biosourced platform chemicals. ACS Sustainable Chem Eng. 2019;7:6471-9.
138. Zhang M, Alferov K, Xiao M, Han D, Wang S, Meng Y. Continuous dimethyl carbonate synthesis from CO2 and methanol using Cu-Ni@VSiO as catalyst synthesized by a novel sulfuration method. Catalysts. 2018;8:142.
139. Lee KM, Jang JH, Balamurugan M, Kim JE, Jo YI, Nam KT. Redox-neutral electrochemical conversion of CO2 to dimethyl carbonate. Nat Energy. 2021;6:733-41.
140. Li X, Han S, Wu W, et al. Convergent paired electrosynthesis of dimethyl carbonate from carbon dioxide enabled by designing the superstructure of axial oxygen coordinated nickel single-atom catalysts. Energy Environ Sci. 2023;16:502-12.
141. Wang H, Wu L, Lan Y, Zhao J, Lu J. Electrosynthesis of cyclic carbonates from CO2 and diols in ionic liquids under mild conditions. Int J Electroch Sci. 2011;6:4218-27.
142. Xiao Y, Chen B, Yang H, Wang H, Lu J. Electrosynthesis of enantiomerically pure cyclic carbonates from CO2 and chiral epoxides. Electrochem Commun. 2014;43:71-4.
143. Zhang J, Shan S, Shi Y, Hou Y, Wang H, Lu J. Highly efficient electrocatalysis for the fixation of CO2 into cyclic carbonates with carbon sphere-loaded copper nanoparticles cathode material. J Electroanal Chem. 2021;882:114962.
144. Pérez-Gallent E, Figueiredo MC, Koper MTM. Mechanistic study of the electrosynthesis of propylene carbonate from propylene oxide and CO2 on copper electrodes. ChemElectroChem. 2019;6:2917-23.
145. Li J, Al-mahayni H, Chartrand D, Seifitokaldani A, Kornienko N. Electrochemical formation of C−S bonds from CO2 and small-molecule sulfur species. Nat Synth. 2023;2:757-65.
146. Wu Y, Zhao J, Zhang B. Electrochemical C−S bond construction from CO2 and an inorganic sulfur source. Sci Bull. 2023;68:1466-8.
147. Pan B, Wang Y, Li Y. Understanding and leveraging the effect of cations in the electrical double layer for electrochemical CO2 reduction. Chem Catal. 2022;2:1267-76.
148. Qin X, Hansen HA, Honkala K, Melander MM. Cation-induced changes in the inner- and outer-sphere mechanisms of electrocatalytic CO2 reduction. Nat Commun. 2023;14:7607.
149. Jin S, Hao Z, Zhang K, Yan Z, Chen J. Advances and challenges for the electrochemical reduction of CO2 to CO: from fundamentals to industrialization. Angew Chem Int Ed Engl. 2021;60:20627-48.
150. Yang Y, Shi Y, Yu H, Zeng J, Li K, Li F. Mitigating carbonate formation in CO2 electrolysis. Next Energy. 2023;1:100030.
151. Govindarajan N, Xu A, Chan K. How pH affects electrochemical processes. Science. 2022;375:379-80.
152. Zhong H, Fujii K, Nakano Y, Jin F. Effect of CO2 bubbling into aqueous solutions used for electrochemical reduction of CO2 for energy conversion and storage. J Phys Chem C. 2015;119:55-61.
153. Overa S, Crandall BS, Shrimant B, et al. Enhancing acetate selectivity by coupling anodic oxidation to carbon monoxide electroreduction. Nat Catal. 2022;5:738-45.
154. Huang JE, Li F, Ozden A, et al. CO2 electrolysis to multicarbon products in strong acid. Science. 2021;372:1074-8.
155. Nguyen DLT, Jee MS, Won DH, Oh H, Min BK, Hwang YJ. Effect of halides on nanoporous Zn-based catalysts for highly efficient electroreduction of CO2 to CO. Catal Commun. 2018;114:109-13.
156. Resasco J, Lum Y, Clark E, Zeledon JZ, Bell AT. Effects of anion identity and concentration on electrochemical reduction of CO2. ChemElectroChem. 2018;5:1064-72.
157. Yuan L, Zeng S, Zhang X, Ji X, Zhang S. Advances and challenges of electrolyzers for large-scale CO2 electroreduction. Mater Rep Energy. 2023;3:100177.
158. Jia S, Zhu Q, Wu H, et al. Efficient electrocatalytic reduction of carbon dioxide to ethylene on copper-antimony bimetallic alloy catalyst. Chin J Catal. 2020;41:1091-8.
159. Feng J, Gao H, Zheng L, et al. A Mn-N3 single-atom catalyst embedded in graphitic carbon nitride for efficient CO2 electroreduction. Nat Commun. 2020;11:4341.
160. Zhao Q, Zhang C, Hu R, et al. Selective etching quaternary MAX phase toward single atom copper immobilized MXene (Ti3C2Clx) for efficient CO2 electroreduction to methanol. ACS Nano. 2021;15:4927-36.
161. Chen Z, Zhang X, Liu W, et al. Amination strategy to boost the CO2 electroreduction current density of M-N/C single-atom catalysts to the industrial application level. Energy Environ Sci. 2021;14:2349-56.
162. Hoang TTH, Verma S, Ma S, et al. Nanoporous copper-silver alloys by additive-controlled electrodeposition for the selective electroreduction of CO2 to ethylene and ethanol. J Am Chem Soc. 2018;140:5791-7.
163. Ren W, Tan X, Jia C, et al. Electronic regulation of nickel single atoms by confined nickel nanoparticles for energy-efficient CO2 electroreduction. Angew Chem Int Ed Engl. 2022;61:e202203335.
164. Gabardo CM, O’brien CP, Edwards JP, et al. Continuous carbon dioxide electroreduction to concentrated multi-carbon products using a membrane electrode assembly. Joule. 2019;3:2777-91.
165. Endrődi B, Samu A, Kecsenovity E, Halmágyi T, Sebők D, Janáky C. Operando cathode activation with alkali metal cations for high current density operation of water-fed zero-gap carbon dioxide electrolyzers. Nat Energy. 2021;6:439-48.
166. Wu Y, Chen C, Yan X, et al. Enhancing CO2 electroreduction to CH4 over Cu nanoparticles supported on N-doped carbon. Chem Sci. 2022;13:8388-94.
167. Xia C, Zhu P, Jiang Q, et al. Continuous production of pure liquid fuel solutions via electrocatalytic CO2 reduction using solid-electrolyte devices. Nat Energy. 2019;4:776-85.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Author Biographies




















Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].