



Use of synthetic communities to study microbial ecology of the gut

 ,
, Abstract
The application of in vitro synthetic microbial communities is an excellent approach to model the ecological interactions between microbes in the human gastrointestinal tract. Although DNA-based studies have provided a wealth of information, they do not consider the ecological properties of the human gut microbiota. Ecological interactions between gut microbes of interest can be studied by applying synthetic communities. This review describes the considerations that should be taken into account when constructing a synthetic community by discussing example research questions that can be answered by using a synthetic microbial community, the choice of microbial species, the growth conditions, possible reactor setups, and the parameters to analyze.
Keywords
INTRODUCTION
The human intestine is home to a complex microbial ecosystem that plays an important role in the conversion of indigested food components and in defending our body against unwanted intruders. Our knowledge on the microbiota, i.e., the collective number of microbes in the intestine, in relation to our health and diet has dramatically increased. This is in part due to explosive technological developments in high-throughput DNA-targeted approaches such as next generation sequencing in the past decades[1,2]. Nowadays, studies that include 16S rRNA gene or metagenome profiles from hundreds to thousands of subjects are no longer an exception. Among other observations, these studies have collectively revealed that the microbiota composition differs between individuals, thereby confirming and extending the observations of individuality of the microbiota based on 16S rRNA gene fingerprinting[3]. In addition, the microbiota composition shows from birth a succession towards a stable diverse adult microbiota composition, which subsequently declines on average in diversity during aging. Remarkably, metagenomics analyses have demonstrated that the functions encoded by the microbiota show high similarities between individuals, which seems to contrast with the individuality at 16S rRNA gene observations[4]. Combined, these observations indicate functional redundancy among various intestinal microbial taxa.
Although these extensive “DNA-based” studies have collectively provided a wealth of information, even within the most deeply phenotyped cohorts, only a small fraction of the compositional diversity within the intestinal microbiota between humans can be explained by factors such as diet and host genetics and physiology[1,2,5]. A recent comment in Nature identifies that many fundamental questions remain unanswered[6]. Part of this can be explained by the vast (micro)biological variation between humans and the fact that most microbiota studies rely on snapshot cross-sectional analyses. For example, a recent study demonstrated that two Dutch pre-diabetic cohorts with nearly identical selection criteria for recruitment showed drastic microbiota differences at baseline[7]. This exemplifies that any cross-sectional comparison between groups of subjects will likely provide microbiota compositional differences, and that cross-sectional comparisons alone are too limited to understand the biological differences underlying the observed variation in composition. In addition, it is very likely that the variation in microbiota composition between individuals is due to the functional redundancy between microbes in combination with the versatility of microbes, which easily change their metabolism depending on the environmental conditions. Indeed, intervention studies targeting the microbiota frequently show a minor impact on microbial composition but a large impact on microbiota activity, suggesting that microbes change their activity as a response to a change in their environment[8,9]. This important ecological characteristic of microbes is not reflected in data generated by DNA-based approaches. Hence, there is a need for microbial ecological approaches that take functional redundancy and versatility of microbes into consideration in order to increase our understanding of the intestinal microbiota. This can be done by in vitro incubation studies in which the activity of microbes can be assessed and controlled.
Numerous in vitro models to study the microbiota in the intestine have been developed and validated. These include, among others, “simple” batch incubations, fermenter systems, complex models simulating different parts of the intestine, and dedicated systems to study host-microbe interactions[10]. These in vitro models can be inoculated with fecal specimens or a selected set of microbes also called a synthetic community. The use of fecal specimens as inoculum has the benefit that the in vitro system uses the original set of microbes from an individual and thereby reflects the closest approximation of their respective microbiota. Several in vitro model studies using fecal inocula have already reflected that the microbiome is highly personalized and influenced by other sources of variation, such as age of the host and intestinal location that are observed in vivo[11-13]. However, the intestine can only be approximated in any in vitro model, and it is evident that the choice of media and conditions results in a selection of microbes that are best adapted to the respective conditions. The use of synthetic communities has the benefit that microbes can be selected as representatives of certain functions within the ecosystem, and that is fruitful for obtaining mechanistic insights into their activity within the ecosystem and how microbes interact with each other. This review focuses on synthetic communities of the human intestine and how these can be used to study the human intestinal ecosystem.
HOW TO CONSTRUCT A SYNTHETIC MICROBIAL COMMUNITY
To construct a synthetic microbial community, it is key to begin with well-defined research questions and hypotheses. This simplifies the experimental design, which includes but is not limited to: (1) the choice of representative microbes; (2) defining the growth conditions; (3) selecting the reactor setup; and (4) deciding the parameters to analyze [Table 1].
Considerations for constructing a synthetic microbial community
Choice of representative microbes | • Natural growth environment • Microbial activities • Genome sequenced • Culturable • In vivo presence and abundance • Compatibility with other microbes of interest • Growth rate in vitro • Number of species |
Defining the growth conditions | • Energy, carbon, and nitrogen source(s) • Nutrients • pH • Redox conditions: oxic/anoxic • Timing of nutrient supply |
Selecting the reactor setup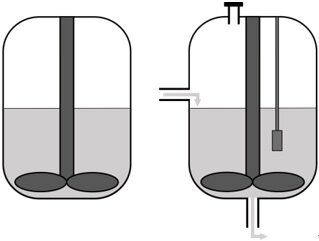 | • Batch • Continuous bioreactor • Mini-bioreactor • Complex model |
Parameters to analyze | • Microbial composition and relative abundance • Transcriptome • Proteome • Metabolome • Tracing substrate • Visualization |
The choice of representative microbes
The choice of which microbes to include in the synthetic community depends on the research question. For example, a synthetic community can be a model of a certain environment, i.e., a specific region of the gut (small or large intestine, lumen, or mucosal layer); a community that performs a certain action, such as the degradation of dietary glycans; a trophic interaction, e.g., cross-feeding on another species’ metabolites; or a disease mechanism, for instance overgrowth of an opportunistic pathogen. Species that are involved in the environment and action of interest can be selected based on whether their genomes have been sequenced, their functional roles in the gut, and their respective in vivo presence and abundance. A sequenced genome is essential for the downstream analyses of the synthetic community, and knowledge about function, presence, and abundance allows a solid design of a community that best represents the in vivo situation of interest. There are multiple datasets available that can aid in the selection of gut species of interest. These include, among others, the Human Microbiome Project, gutMDisorder, MetaHit, and the Unified Human Gastrointestinal Genome[11,14-16]. An overview of cultured species can be found in databases such as
Defining the growth conditions
The conditions in which the synthetic microbial community will grow must be defined. For example, for modeling the gut microbiota in the colon, the culture environment should be anoxic, which allows growth of gut anaerobes. This can be achieved by gas exchanging the head space of the reactor or flask that is used to the desired gas phase and including a suitable reducing agent in the medium. Furthermore, the medium should contain a buffer or continuous automatic pH control, which should warrant a stable pH during experiments to ensure reproducibility. The pH should be set to reflect the environment that is modeled in the synthetic community and allow the selected species to thrive. Another consideration is the choice of the medium to use for the synthetic community. There are complex media available that mimic a selected part of the intestinal tract, such as simulated-ileal-environment medium and simulated-colonic-environment medium[20,21]. Alternatively, a basal-defined medium creates a more controlled system[22]. Next, the sources of energy, carbon, and nitrogen can be selected. This choice largely depends on the research questions because there are many potential substrates in the gut, such as dietary carbohydrates and proteins, host-derived mucin glycans in general, or human milk oligosaccharides in the infant gut. Depending on the location in the gut, the function of interest, and the species present in the synthetic microbial community, one or multiple of these substrates can be added to the medium. To incorporate dietary components as nutrients for the synthetic community, it should be considered that a diet is highly variable, in terms of both timing and composition. In vivo, dietary components are not constantly available to the gut microbiota. Therefore, dietary nutrients can be added to the synthetic community at regular intervals, instead of providing a constant supply[23]. Furthermore, the composition of dietary nutrients should be considered carefully, as there is large inter- and intra-individual variation in food intake[24]. Diet greatly influences the composition of the gut microbiota, leading to variations in its composition, for example by geographical location; by varying intakes of fibers, fat, protein, and carbohydrates; and by a predominantly vegetarian or carnivorous diet[25]. A synthetic community can be constructed with the aim to cover as many (major) functions of the in vivo human gut microbiota as possible. For this approach, a complex medium is required. For example, one experiment consisted of a bioreactor which was provided with a constant flow of mucin and acetate, and the dietary components pectin, xylan, starch, and inulin were added three times per day to mimic a human diet to construct a minimal microbiome that covered key ecological and metabolic properties of the human gut microbiota[23]. Alternatively, the focus of the synthetic community can be on a single function of the human gut microbiota. For example, mucin degradation was studied by growing mucin-degrading bacteria together with their syntrophic partners in a medium containing mucin as the sole carbon source[26,27].
Selecting the reactor setup
The next consideration is the in vitro system in which the synthetic microbial community will grow. The community can be grown in batch in flasks, which is a straightforward method to study interactions in a simple microbial community under controlled conditions. However, batch cultivation leads to quick depletion of substrates and rapid buildup of potentially toxic metabolites and is more suited to studying short-term community dynamics and assaying the end-products of fermentation. Chemostat bioreactors have been and continue to be successfully used to culture complex microbial communities found in feces[28] and simpler defined bacterial communities[29]. These systems provide insight into compositional and metabolic changes in microbial communities over time and in response to perturbations[30]. A chemostat bioreactor with controlled inflow of fresh medium and outflow of waste, pH, or redox potential control is a more complicated method compared to batch culturing but allows the study of more complex microbial interactions. However, compared to batch experiments, the number of comparisons between conditions or inocula that can be made is limited.
It is important to consider the growth rate of the microbes for the refreshing rate of the medium. Specifically, a constant flow of fresh nutrients can be applied, or pre-defined feed inputs at set times to mimic the host diet can be employed. For more high-throughput analyses, for example when multiple conditions need to be tested, mini-bioreactors can be applied[31]. Alternatively, a specific part of the gut can be modeled, such as the different regions of the intestinal tract, the mucus layer, or the lumen. For a more complete model of the intestinal tract, a more complex model such as mucosal-simulator of human intestinal microbial ecosystem (M-SHIME) can be applied. The M-SHIME model incorporates both a luminal and a mucosal compartment. Another example of a more complex model is the TNO in vitro model of the colon (TIM-2). TIM-2 models the colon by applying peristaltic movements and a dialysis system for the removal of metabolites[32]. Similarly, a recent system has been described to simulate the human ileum[33]. Furthermore, the SIMGI (simulator gastro-intestinal) model consists of a gastric component, a small intestine component, and three components that represent the different regions of the colon[34].
The ultimate choice of which reactor set-up to apply for studying the synthetic microbial community of interest depends, among others, on the complexity of the community, the gut environment of interest, the research question(s), the desired output, and the availability of resources. For example, a batch culture system could be chosen when the impact of multiple food components is compared and contrasted, while a continuous culture setup would be the preferred choice when the response of the community after an intervention is the focus of the study.
Deciding the parameters to analyze
It is crucial to define what output needs to be measured during growth of the synthetic microbial community to answer the research questions. This can include measurements of relative and/or absolute abundances, the decrease of substrate, the increase of metabolites, transcriptomics, and proteomics. Table 2 lists potential output of the synthetic community and examples of associated techniques.
Potential output of a synthetic community and examples of tools that can be applied to measure these outputs
| Output | Technique | Advantages | Disadvantages |
| Microbial composition and relative abundance | 16S rRNA gene sequencing | Straightforward method and large database of sequences available allowing highly accurate comparisons | Only provides relative abundances* Species-level profiling not reliable |
| Internal transcribed spacer sequencing | Species-level microbial profiling | Only provides relative abundances* | |
| Quantitative PCR | Provides relative and absolute abundances | Only possible when specific primers are available | |
| Transcriptome | RNA sequencing | Not limited by the design of probes | Requires specific bioinformatic knowledge to handle large datasets |
| Microarray | Examine the expression of thousands of genes simultaneously | Requires reference genomes for the organisms of interest to design probes | |
| Proteome | Immunoassays (e.g., enzyme-linked immunosorbent assay) | Detection of proteins with a low abundance | Targets specific proteins |
| Gel-based protein separation (e.g., sodium dodecyl sulfate-polyacrylamide gel electrophoresis) | Suitable for complex samples | Downstream analysis method required Labor intensive | |
| Chromatography-based protein separation | Suitable for complex samples | Downstream analysis method required | |
| Protein microarray | High throughput Suitable for complex samples | The antibodies need to be predefined | |
| Mass spectrometry | High throughput Sensitive Suitable for complex samples | Costly; specific analytical expertise needed | |
| Metabolome/metabonome | High-performance liquid chromatography (HPLC) | Quick, automated, and accurate method to identify certain chemical components in a sample | Costly; several systems and columns needed for different chemical categories |
| Liquid chromatography-mass spectrometry (LC-MS) | Separation of molecules (HPLC) combined with structural identification of individual compounds with high detection sensitivity and molecular specificity (MS) | Costly; not all mobile phases used for HPLC are compatible with MS | |
| Gas chromatography | Quick, automated, and accurate method to identify certain chemical components in a sample | Several systems and columns needed for different chemical categories | |
| In vitro nuclear magnetic resonance | Monitoring microbial metabolism; provides information about molecular structure at atomic level; pathway reconstruction | Only small molecules can be detected | |
| Tracing substrate | DNA or RNA stable-isotope probing | Trace which community member incorporates a certain component | Careful experiment design, the required sensitivity, the approach selected for separating labeled nucleic acid biomarkers, and the downstream analysis intended for the labeled material |
| In vitro nuclear magnetic resonance | Monitoring microbial metabolism; provides information about molecular structure at atomic level | Rather low sensitivity | |
| Bioorthogonal non-canonical amino acid tagging combined with fluorescently activated cell sorting and sequencing[35] | Identification of translationally active microorganisms in the community | New method, so requires further optimization and validation, in particular for application to synthetic microbial communities | |
| Visualization | Fluorescence in situ hybridization (FISH) | Visualization of localization of community members | Limited by the availability of fluorescent probes No quantification Detection limit |
| Microautoradioactivity combined with FISH | Identification of metabolically active microorganisms in mixed communities | Low rRNA copy numbers are undetectable with standard FISH techniques |
CONCLUSION
Synthetic microbial communities are a great tool to study ecological interactions between gut microbes of interest. They can provide knowledge on microbial functionality beyond that derived from DNA-based techniques. Studies using synthetic communities have proven valuable for doing gut microbiota-related research in uncovering cross-feeding actions and collaborative resource sharing. For example, a bioreactor supplemented with dietary fibers and mucin was applied to study trophic interactions and niche occupation between microbes in vitro[23]. The application of synthetic microbial communities still has several limitations. First, not all gut microbes have been cultured or have their genome sequence determined, which could result in a lack of function in the synthetic community. Second, even though synthetic communities are designed to mimic the natural environment as well as possible, microbes could adopt different roles than the ones they have in vivo. As such, host factors such as hormones and immune factors are usually either not or poorly represented in in vitro models. Third, tracing molecules in a synthetic community is difficult, as it is not possible to distinguish which species are using a certain substrate, or which species produce a certain metabolite.
However, to move forward and provide functional data on microbial ecosystems, we are convinced that synthetic communities are the way to go, and this review provides a workflow on how to conduct qualitative experiments when doing so. We provide examples of how a synthetic minimal microbiome can be applied to answer research questions of interest. We see great potential, for instance when the goal of the research is to achieve a certain functional output, such as the one described for optimizing butyrate production[18]. Metabolic modeling can aid the design of synthetic communities, as metabolic models can predict functions, interactions, and responses to condition changes, but also point towards knowledge gaps. Therefore, in vitro synthetic communities and metabolic modeling can complement each other[37,38]. Future research on synthetic communities for modeling the gut microbiota holds great potential for new insight into microbial actions and functionality that impact the gut system. Moreover, advances in synthetic biology will certainly help in investigating yet unknown gut microbial functions observed by metagenomics[39].
DECLARATIONS
Authors’ contributionsConceptualization: Zoetendal EG, Belzer C
Wrote the manuscript: Berkhout MD, Zoetendal EG, Plugge CM, Belzer C
Reviewed and edited the manuscript: Zoetendal EG, Plugge CM, Belzer C
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Netherlands Ministry of Education, Culture and Science and the Dutch Research Council (NWO) (SIAM Gravitation grant, project 024.002.002).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Falony G, Joossens M, Vieira-Silva S, et al. Population-level analysis of gut microbiome variation. Science 2016;352:560-4.
2. Rothschild D, Weissbrod O, Barkan E, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018;555:210-5.
3. Zoetendal EG, Akkermans AD, De Vos WM. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol 1998;64:3854-9.
4. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature 2012;489:220-30.
5. Zhernakova A, Kurilshikov A, Bonder MJ, et al. LifeLines cohort study. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016;352:565-9.
7. Hermes GDA, Reijnders D, Kootte RS, et al. Individual and cohort-specific gut microbiota patterns associated with tissue-specific insulin sensitivity in overweight and obese males. Sci Rep 2020;10:7523.
8. O'Keefe SJ, Li JV, Lahti L, et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat Commun 2015;6:6342.
9. Salonen A, de Vos WM. Impact of diet on human intestinal microbiota and health. Annu Rev Food Sci Technol 2014;5:239-62.
10. Elzinga J, van der Oost J, de Vos WM, Smidt H. The use of defined microbial communities to model host-microbe interactions in the human gut. Microbiol Mol Biol Rev 2019;83:e00054-18.
11. Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012;486:207-14.
12. Auchtung JM, Robinson CD, Britton RA. Cultivation of stable, reproducible microbial communities from different fecal donors using minibioreactor arrays (MBRAs). Microbiome 2015;3:42.
13. Reichardt N, Vollmer M, Holtrop G, et al. Specific substrate-driven changes in human faecal microbiota composition contrast with functional redundancy in short-chain fatty acid production. ISME J 2018;12:610-22.
14. Cheng L, Qi C, Zhuang H, Fu T, Zhang X. gutMDisorder: a comprehensive database for dysbiosis of the gut microbiota in disorders and interventions. Nucleic Acids Res 2020;48:D554-60.
15. Qin J, Li R, Raes J, et al. MetaHIT Consortium. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010;464:59-65.
16. Almeida A, Nayfach S, Boland M, et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol 2021;39:105-14.
17. Söhngen C, Podstawka A, Bunk B, et al. BacDive--the bacterial diversity metadatabase in 2016. Nucleic Acids Res 2016;44:D581-5.
18. Clark RL, Connors BM, Stevenson DM, et al. Design of synthetic human gut microbiome assembly and butyrate production. Nat Commun 2021;12:3254.
19. Zimmermann J, Kaleta C, Waschina S. gapseq: informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. Genome Biol 2021;22:81.
20. Beumer R, de Vries J, Rombouts F. Campylobacter jejuni non-culturable coccoid cells. Int J Food Microbiol 1992;15:153-63.
21. Müsken A, Bielaszewska M, Greune L, et al. Anaerobic conditions promote expression of Sfp fimbriae and adherence of sorbitol-fermenting enterohemorrhagic Escherichia coli O157:NM to human intestinal epithelial cells. Appl Environ Microbiol 2008;74:1087-93.
23. Shetty SA, Kostopoulos I, Geerlings S, Smidt H, de Vos WM, Belzer, C. Minimalist approach for deciphering the ecophysiology of human gut microbes. In: Kostopoulos I, editor. Mucin and human milk oligosaccharides utilization: a strategy of Akkermansia muciniphila to ensure survival in the human gut. Wageningen; 2021. p. 131-76.
24. Johnson AJ, Vangay P, Al-Ghalith GA, et al. Personalized Microbiome Class Students. Daily sampling reveals personalized diet-microbiome associations in humans. Cell Host Microbe 2019;25:789-802.e5.
25. Bibbò S, Ianiro G, Giorgio V, et al. The role of diet on gut microbiota composition. Eur Rev Med Pharmacol Sci 2016;20:4742-9.
26. Belzer C, Chia LW, Aalvink S, et al. Microbial metabolic networks at the mucus layer lead to diet-independent butyrate and vitamin B12 production by intestinal symbionts. mBio 2017;8:e00770-17.
27. Bunesova V, Lacroix C, Schwab C. Mucin cross-feeding of infant bifidobacteria and eubacterium hallii. Microb Ecol 2018;75:228-38.
28. Yen S, McDonald JA, Schroeter K, et al. Metabolomic analysis of human fecal microbiota: a comparison of feces-derived communities and defined mixed communities. J Proteome Res 2015;14:1472-82.
29. Drake DR, Brogden KA. Continuous-culture chemostat systems and flowcells as methods to investigate microbial interactions. In: Brogden KA, Guthmiller JM, editors. Polymicrobial diseases. Washington: ASM Press; 2002. p. 21-30.
30. Newton DF, Macfarlane S, Macfarlane GT. Effects of antibiotics on bacterial species composition and metabolic activities in chemostats containing defined populations of human gut microorganisms. Antimicrob Agents Chemother 2013;57:2016-25.
31. O’Donnell MM, Rea MC, Shanahan F, Ross RP. The use of a mini-bioreactor fermentation system as a reproducible, high-throughput ex vivo batch model of the distal colon. Front Microbiol 2018;9:1844.
32. Venema K. The TNO in vitro model of the colon (TIM-2). In: Verhoeckx K, Cotter P, López-expósito I, Kleiveland C, Lea T, Mackie A, Requena T, Swiatecka D, Wichers H, editors. The impact of food bioactives on health. Cham: Springer International Publishing; 2015. p. 293-304.
33. Stolaki M, Minekus M, Venema K, et al. Microbial communities in a dynamic in vitro model for the human ileum resemble the human ileal microbiota. FEMS Microbiol Ecol 2019;95:fiz096.
34. Barroso E, Cueva C, Peláez C, Martínez-cuesta MC, Requena T. The computer-controlled multicompartmental dynamic model of the gastrointestinal system SIMGI. In: Verhoeckx K, Cotter P, López-expósito I, Kleiveland C, Lea T, Mackie A, Requena T, Swiatecka D, Wichers H, editors. The impact of food bioactives on health. Cham: Springer International Publishing; 2015. p. 319-27.
35. Taguer M, Shapiro BJ, Maurice CF. Translational activity is uncoupled from nucleic acid content in bacterial cells of the human gut microbiota. Gut Microbes 2021;13:1-15.
36. Galazzo G, van Best N, Benedikter BJ, et al. How to count our microbes? Front Cell Infect Microbiol 2020;10:403.
37. Vrancken G, Gregory AC, Huys GRB, Faust K, Raes J. Synthetic ecology of the human gut microbiota. Nat Rev Microbiol 2019;17:754-63.
38. D’hoe K, Vet S, Faust K, et al. Integrated culturing, modeling and transcriptomics uncovers complex interactions and emergent behavior in a three-species synthetic gut community. Elife 2018;7:e37090.
Cite This Article
 , ... Clara Belzer
, ... Clara BelzerHow to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright














Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].