



Transcriptomic characterization of Wolbachia endosymbiont from Leuronota fagarae (Hemiptera: Psylloidae)
 0
0  , , ...
, , ... Abstract
Aim:Wolbachia species are among the most abundant intracellular endosymbionts of insects worldwide. The extensive distribution of Gram-negative Wolbachia among insects highlights their evolutionary success and close relationship with many insect host species. This study aimed to characterize a novel Wolbachia strain from the Wild Lime Psyllid, Leuronota fagarae (L. fagarae), to understand its evolutionary relationship with Wolbachia from psyllid pests like Diaphorina citri, the vector of Huanglongbing (HLB).
Methods: Wild-caught L. fagarae colonies from Florida, USA, were maintained on Zanthoxylum fagara. RNA was extracted from the salivary glands, heads, and whole bodies of male and female adult L. fagarae. Four cDNA libraries were sequenced using short read technology and de novo transcriptome assembly was performed. Multilocus sequence typing (MLST) of nine conserved loci and wsp gene analysis classified the strain’s phylogeny, while sequence mapping and functional annotation provided insight into host-microbe interactions.
Results: The new Wolbachia strain, designated Wolbachia endosymbiont of Leuronota fagarae (wLfag-FL), was assigned to supergroup B, showing relation to Wolbachia strains of other related psyllids. Transcriptome analysis identified 1,359 Wolbachia transcripts with 465 assigned functions encompassing metabolic and secretion system pathways. Ankyrin domain proteins and a partial bacterioferritin sequence were detected, suggesting nutritional provisioning roles.
Conclusion: The characterization of wLfag-FL expands the known Wolbachia host range and informs HLB-related pest biology. Its phylogenetic placement and transcript annotations offer insights into symbiotic interactions, potentially guiding environmentally safe pest control strategies targeting psyllid fitness and pathogen transmission.

Keywords
INTRODUCTION
The Wild Lime Psyllid, Leuronota fagarae (L. fagarae) Burckhardt (Hemiptera: Psylloidea), is an invasive species of Florida, USA, with origins traced to Paraguay, South America[1]. The first report of L. fagarae in the United States occurred in southern Florida when adults were discovered feeding and reproducing on citrus relative Zanthoxylum fagara (L.) Sarg. (Sapindales: Rutaceae)[1]. The 2001 discovery of L. fagarae in southern Florida led to the first classification of the species since its original description in 1988 from two locations in Paraguay[2]. Rolled leaf edges that provide safeguards for developing nymphs indicate a typical phenotype of L. fagarae feeding on citrus[1]. In Florida, the habitat range of L. fagarae overlaps with the Asan Citrus Psyllid (ACP, Diaphorina citri Kuwayama) in the transition zone from non-cultivated areas to areas of cultivated citrus trees. This proximity, as well as taxonomic relationship and potentially shared food source, provides an opportunity for the transfer of intracellular microbes through plant feeding[3], such as Wolbachia spp.[4].
The parasitism-mutualism balance linking host and endosymbionts is a major influence on systemic biological processes within an organism[5]. Previous studies have determined that endosymbionts influence fundamental aspects of host insect biology, including nutritional mechanisms[6], reproduction[7], tolerance of abiotic conditions[8,9], and the reduction of an insect’s ability to vector pathogens[10]. Molecular pathways critical to the health of endosymbionts and insect hosts provide targets of interest for designing therapeutics to induce phenotypic changes in the insect. Additionally, symbiotic relationships are of interest in evolutionary genetics, as codependence can result in genetic markers in endosymbiont genomes, which inform the evolutionary history and adaptation of the insect[11,12]. Insect diets deficient in vitamins, sterols, and essential amino acids[13] are supplemented by symbiotic microorganisms that produce the necessary dietary nutrients[14]. Sap ingesting psyllids, such as L. fagarae, are hosts to a variety of bacterial endosymbionts, most notably Gram-negative Wolbachia of the order Rickettsiales, which are currently found in 40%-60% of insect species[15,16]. Our characterization of Wolbachia in L. fagarae is specifically noteworthy due to Wolbachia’s potential to modulate the fitness of disease-vectoring insects[17] and its close relationship to the Wolbachia strain found in the Huanglongbing (HLB) vector Diaphorina citri. Previous analysis of Wolbachia genome assemblies further determined that these microorganisms are without complete sets of metabolic pathways present in closely related, nonsymbiotic bacteria of the order Rickettsiales[18], suggesting that Wolbachia can manipulate host metabolic pathways[19,20]. This dynamic influence, along with the exclusive vertical transmission through maternal hosts, contributes to the exploitation of major reproductive influences through Wolbachia-host relationships[21].
Wolbachia are currently classified into separate monophyletic lineages, or supergroups, denoted A to S[22]. Here, we looked to classify the evolutionary placement of Wolbachia endosymbiont of Leuronota fagarae (wLfag-FL) in the supergroup classification system and to examine the transcriptome of wLfag-FL to uncover conserved or unique transcripts informing the symbiotic relationships of Wolbachia and psyllid hosts. Our study uses an multilocus sequence typing (MLST) of nine loci to classify wLfag-FL phylogeny and leverages published Wolbachia genomes from D. citri to perform a comparative analysis of Wolbachia loci found in two psyllid species invasive to Florida, USA. L. fagarae presents an important organism to study in parallel to D. citri, as recent research on the Australian finger lime produced a novel line of stable antimicrobial peptides (SAMPs) to bolster citrus immunity against CLas infection. Since L. fagarae feeds on the citrus relative wild lime, is closely related to D. citri, and is invasive to Florida, the biology of L. fagarae and its endosymbionts informs HLB research and may reveal more naturally occurring therapeutic molecules to combat HLB. A thorough comparative investigation of the wLfag-FL transcriptome was not possible due to the low number of transcripts recovered from wLfag-FL, although transcripts encoding proteins essential for nutrient provisioning, proteins with secretory domains, and proteins with ankyrin repeat (ANK) domains characteristic to Wolbachia were identified to inform host-microbe interactions.
METHODS
L. fagarae collection
Colonies of wild-caught L. fagarae found on Zanthoxylum fagara were maintained at the University of Florida Southwest Florida Research & Education Center in Immokalee, FL, U.S.A., for 5 years. L. fagarae colonies were maintained on citrus relative Zanthoxylum fagara (L.) Sarg (Sapindales: Rutaceae) by the Qureshi lab and sample collection from the colony occurred in 2017. After collection, psyllids were immediately placed into TRIzolTM-LS Reagent (InvitrogenTM #10296028, 200 mL) for RNA purification[23].
RNA extraction, cDNA preparation, and library sequencing
Four cDNA libraries were made from RNA extracted from salivary glands, heads, or whole-body tissue of L. fagarae. Both cDNA libraries from the salivary gland and head tissues were obtained from combined male and female samples, while both whole-body cDNA libraries were made from either male or female psyllids. In total, 900 psyllids were collected to build cDNA datasets where 500 psyllids were dissected for the mixed salivary gland dataset, 380 psyllids were dissected for the mixed heads dataset, and 10 psyllids each, male and female, were used for the whole-body dataset RNA extractions. RNA was purified using the Direct-zol RNA MicroPrep method for the adult whole body and mixed heads extractions, and TRIzol LS, Na-Acetate/ethanol precipitation was used to purify the mixed salivary glands extractions. Total RNA concentration dissolved in nuclease-free water was assessed with the Qubit BR assay. The results showed that the mixed salivary gland extractions yielded 2.8 ng/μL of RNA, the mixed head extractions yielded 4.4 ng/μL of RNA, adult males yielded 30.8 ng/μL of RNA, and adult females yielded 134 ng/μL of RNA. To sequence the transcriptome of each tissue or whole psyllid, we used the NEB Ultra II Directional RNAseq Library Prep Kit from Illumina paired with the Illumina NextSeq 500 Mid mode throughput 2X150 method. Adapters and low-quality reads (Q < 20) were removed at the University of Florida Interdisciplinary Center for Biotechnology Research (UF|ICBR). Raw reads have been deposited into the Sequence Read Archive under BioProject PRJNA789049 and assembled transcripts are found in BioSample SAMN46999246.
Data processing and sequence identification
De novo assemblies of four resulting cDNA libraries (salivary glands, heads, whole-body males, and whole-body females) were performed using the TRINITY RNA-seq assembler v1.8.0[24] with default parameters. For the initial characterization of Wolbachia sequences, nine local databases containing conserved Wolbachia housekeeping genes: 16S rRNA, wsp, coxA, fbpA, ftsZ, gatB, gltA, groEL, and hcpA were built from transcripts available on NCBI. The Trinity assembled transcripts were aligned to each of the nine local databases using BLASTn. The longest transcripts matching a subject with the lowest E-value scores were selected for MLST. Phylogenetic analysis was performed using a concatenated form of these conserved sequences for each of the samples listed in Table 1. As a result of the MLST study, the three most closely related Wolbachia endosymbionts to wLfag-FL: Spodoptera picta strain Spic_B (CP067976.1), Diaphorina citri strain dawsonii (CP051608.1), and Laodelphax striatellus strain wStri (NZ_MUIX01000001.1) were used as reference genomes for HISAT2 v2.0.4 mapping with default parameters[25] to identify Wolbachia sequences in the four TRINITY assembled datasets. The resulting sequences were subject to BLASTn analysis using the NCBI nt database to verify sequence identity across a broader database. If sequences did not find a match to NCBI via nucleotide, BLASTx was used instead to compare protein homology. BUSCO v5.2.2 was used to assess the transcriptome completeness of obtained Wolbachia orthologs[26]. The CAP3 long read assembler in UGENE v40[27] was used to further assemble fragmented Wolbachia sequences. The contigs that did not assemble any further were grouped with the CAP3 contig assemblies, which were subject to gene annotation.
List of all Wolbachia strains and accessions used in phylogenetic analysis
| Host name | Common name | Taxonomy (order: family) | Strain | Supergroup | Abbreviation | GenBank accession |
| Anastrepha fraterculus | South American fruit fly | Diptera: Tephritidae | clone 46.1 | A | wAfra† | KC589027.1 |
| Carposina sasakii | Peach fruit moth | Lepidoptera: Carposinidae | wCauA | A | wCsas | NZ_CP041215.1 |
| Ceratosolen solmsi | Fig wasp | Hymenoptera: Agaonidae | MIAOwCsol | A | wCsol | CP054598.1 |
| Drosophila ananassae | Fruit fly | Diptera: Drosophilidae | wAna | A | wDana | CP042904.1 |
| Drosophila incompta | Fruit fly | Diptera: Drosophilidae | wInc Cu | A | wDinc | NZ_CP011148.1 |
| Drosophila melanogaster | Fruit fly | Diptera: Drosophilidae | wMelpop | A | wDmel | CP046921.1 |
| Drosophila simulans | Fruit fly | Diptera: Drosophilidae | wAu | A | wDsim1 | CP069055.1 |
| Hypothenemus hampei | Coffee berry borer | Coleoptera: Curculionidae | H control 1 | A | wHham† | KX436087.1 |
| Wiebesia pumilae | Fig wasp | Hymenoptera: Agaonidae | MIAOwWpum | A | wWpum | CP054557.1 |
| Aedes albopictus | Tiger mosquito | Diptera: Culicidae | wAlbB | B | wAalb | CP031221.1 |
| Bactericera cockerelli | Potato psyllid | Hemiptera: Triozidae | clone 3 | B | wBcoc† | KM267307.1 |
| Bemisia tabaci | Silverleaf whitefly | Hemiptera: Aleyrodidae | China 1 | B | wBtab | NZ_CP016430.1 |
| Culex quinquefasciatus | Southern house mosquito | Diptera: Culicidae | wPipPel | B | wCqui | AM999887.1 |
| Diaphorina citri | Asian citrus psyllid | Hemiptera: Liviidae | dawsonii | B | wDcit1 | CP051608.1 |
| Diaphorina citri | Asian citrus psyllid | Hemiptera: Liviidae | KPSwDI10P38 | B | wDcit2 | CP051265.2 |
| Diaphorina citri | Asian citrus psyllid | Hemiptera: Liviidae | Beihai | B | wDcit3† | GQ385974.1 |
| Diaphorina citri | Asian citrus psyllid | Hemiptera: Liviidae | Shenzhen | B | wDcit4† | GU480072.1 |
| Drosophila mauritiana | Fruit fly | Diptera: Drosophilidae | wMau | B | wDmau | CP034335.1 |
| Drosophila simulans | Fruit fly | Diptera: Drosophilidae | wMa | B | wDsim2 | CP069054.1 |
| Homalodisca coagulata | Glassy-winged sharpshooter | Hemiptera: Cicadellidae | GWSS | B | wHcoa† | DQ450164.1 |
| Homalodisca elongata | Glassy-winged sharpshooter | Hemiptera: Cicadellidae | B21HE | B | wHelo† | DQ450154.1 |
| Laodelphax striatellus | Small brown planthopper | Hemiptera: Delphacidae | wStri | B | wLstr | NZ_MUIX01000001.1 |
| Nilaparvata lugens | Brown planthopper | Hemiptera: Delphacidae | wLug | B | wNlug | NZ_MUIY01000001.1 |
| Spodoptera picta | Lily caterpillar | Lepidoptera: Noctuidae | Spic B | B | wSpic | CP067976.1 |
| Brugia pahangi | Filarial nematode worm | Rhabditida: Onchocercidae | FR3 | D | wBpah | CP050521.1 |
| Litomosoides sigmodontis | Filarial nematode worm | Rhabditida: Onchocercidae | wLsig | D | wLsig | CP046577.1 |
| Folsomia candida | Springtail | Collembola: Isotomidae | Berlin | E | wFcan | NZ_CP015510.2 |
| Cimex lectularius | Bed bug | Hemiptera: Cimicidae | wCle | F | wClec | AP013028.1 |
Phylogenetic characterization
Two methods were employed to characterize the evolutionary lineage of Wolbachia found in L. fagarae from Florida. The first approach used a concatenated sequence of nine conserved genes, where sequences were only gathered and analyzed if they were part of a fully assembled Wolbachia genome on NCBI. The resulting concatenated sequence alignment, performed with MUSCLE in UGENE v40, totaled 7,655 nucleotides in length, where partial sequences of CoxA, ftsZ, gatB, gltA, groEL, and hcpA were used along with full-length sequences of fbpA, wsp, and 16S rRNA. The second approach used a portion of the wsp gene that encodes a transmembrane structure, which plays a role in controlled cell death, cell proliferation, pathogenicity, and host immune response[28]. The wsp sequences obtained from NCBI, belonging to various Wolbachia strains of interest, were aligned with MUSCLE in UGENE v40, and trimmed to the limiting sequence, resulting in a length of 609 nucleotides. Both phylogenetic trees were built using a bootstrap value of 1000 and the default Maximum Likelihood parameters in UGENE v40.
Transcriptome functional annotation
Four Wolbachia gene datasets available in the KEGG Automatic Annotation Server (KAAS v2.1) were used to obtain KEGG Orthology identifiers of wLfag-FL sequences[29-31]. The four datasets added together totaled 4,425 sequences and were entitled Wolbachia endosymbiont of Drosophila melanogaster (wol), Wolbachia sp. wRi (wri), Wolbachia endosymbiont of Culex quinquefasciatus Pel (wpi), and Wolbachia endosymbiont strain TRS of Brugia malayi (wbm). Both unassembled and CAP3 assembled sequences were placed into the KAAS server for annotation via the BLAST method and SBH approach. Identification of sequences containing ankyrin domains was carried out post CAP3 assembly of identified wLfag-FL contigs and was initiated by BLASTx analysis to a dataset of 182 Multispecies Wolbachia ankyrin domain-containing proteins from NCBI. Aligned sequences were then verified by selecting whole ExPASy sequence translations[32] of the same frames, which had homology to Wolbachia ANK domain proteins predicted by BLASTx. The translated sequences were compared against the UniProt database for final confirmation.
RESULTS
Phylogenetic analysis
Both phylogenetic characterization strategies using MLST [Figure 1] and wsp [Figure 2] assigned Wolbachia strain wLfag-FL to supergroup B, which contains closely related psyllids along with other Hemipterans, Dipterans, and a Lepidopteran. A wsp gene coding for a surface protein of Wolbachia was specifically chosen for further analysis as it evolves at a much faster rate than other genes such as 16S or ftsZ, and has been previously used in characterizing Wolbachia strains[33]. Moreover, the wsp gene is allotted for further analysis of strains without a completely assembled genome. Analysis was attempted with a protein translation of the wsp gene, but given the lack of whole sequence availability for other Wolbachia strains, incomplete protein translations could not differentiate lineages of closely related strains. The Wolbachia endosymbiont of B. cockerelli is seemingly the closest relative of wLfag-FL according to wsp analysis. All sequence information used to build the phylograms is recorded in Table 1. A depiction of the wsp alignment created in Jalview v2.11.1[34] is available in Supplementary Figure 1.

Figure 1. Unrooted Maximum Likelihood tree for Wolbachia MLST concatenated sequences supporting placement of wLfag-FL in Supergroup B. MLST: Multilocus sequence typing; wLfag-FL: Wolbachia endosymbiont of Leuronota fagarae.

Figure 2. Unrooted Maximum Likelihood tree for Wolbachia wsp sequences highlighting the placement of wLfag-FL in Supergroup B with a distinct evolutionary lineage. wLfag-FL: Wolbachia endosymbiont of Leuronota fagarae.
Transcriptome analysis
Of the 1,359 transcripts mapped to reference Wolbachia genomes by HISAT2, only six sequences did not match Wolbachia through BLASTn to NCBI nt; however, these 6 did match Wolbachia sequences through BLASTx to NCBI nr with the greatest E-value reported as 4e-08. To assess transcriptome completeness of the assembled Wolbachia cDNA sequences, Benchmark Universal Single Copy Ortholog (BUSCO) v5.2.2 was used[26]. In total, 1,332 non-redundant transcripts were compared against the rickettsiales_odb10 database, using transcriptome mode. BUSCO calculated only 37 Complete BUSCOs (10.7%), 14 Fragmented BUSCOs (4.1%), and 294 Missing BUSCOs (85.2%). Seqkit v0.16.1[35] was used to calculate a GC content of 34.8% and an N50 value of 348 nt for Trinity assembled Wolbachia transcripts. When aligning the wLfag-FL transcripts to the annotated sequences of wDcit dawsonii and the wSpic, we identified 634 and 560 alignments to unique transcripts, respectively, and revealed, on average, a 45% annotation rate of our wLfag-FL Trinity transcripts. Roughly 2.5 times more Wolbachia sequences were recovered from the female L. fagarae dataset compared to orthologs recovered in the male dataset. To visualize sequence comparisons between wLfag-FL and other closely related Wolbachia strains, we used AliTv[36] and the Blast Ring Image Generator (BRIG)[37]. An AliTv alignment mapping figure was generated with concatenated wLfag-FL transcripts, post CAP3 assembly, and Wolbachia reference genomes Spodoptera picta Spic B and Diaphorina citri dawsonii [Figure 3]. To generate the BRIG image [Figure 4], Supergroup B Wolbachia genomes from the MLST phylogeny were reused.
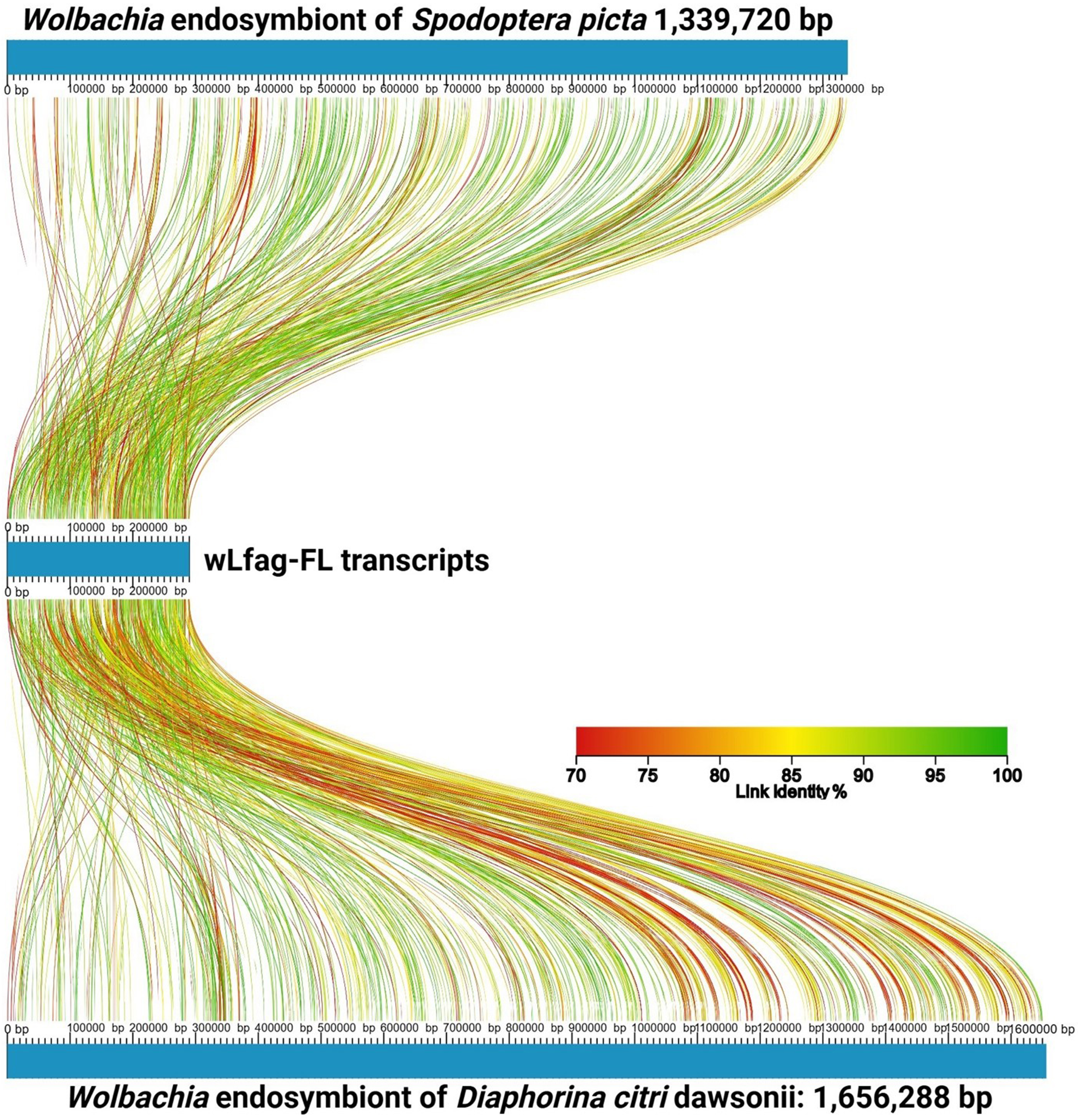
Figure 3. AliTV linear alignment map of wLfag-FL concatenated transcripts mapped to closely related Wolbachia genomes of wDcit1 and wSpic. Accession numbers of full genome references are in Table 1. wLfag-FL: Wolbachia endosymbiont of Leuronota fagarae; wDcit1: Wolbachia endosymbiont of D. citri dawsonii; wSpic: Wolbachia endosymbiont of S. picta.

Figure 4. BRIG alignment of 11 Wolbachia endosymbiont genomes belonging to Supergroup B used in the MLST phylogenetic classification of wLfag-FL. Wolbachia endosymbiont of S. picta was set as the reference genome (red); the outermost ring (dark green) is made of wLfag-FL transcripts. Genome assembly lengths are in parenthesis next to the names of the Wolbachia strain. Genome accessions are in Table 1. BRIG: Blast Ring Image Generator; MLST: multilocus sequence typing; wLfag-FL: Wolbachia endosymbiont of Leuronota fagarae.
Functional annotation
As a result of the KEGG ontology analysis, 465 transcripts were assigned a K number, of which 279 were unique of the 828 total sequences examined. The map pathway function available in the KO Database was used to group the unique K number assignments into their associated pathways. The KEGG Orthology Metabolic pathway (ko01100) contained the highest number of assignments with 108 sequences. The bar graph [Figure 5] depicts an abundance of 13 KEGG Orthology pathways containing at least 10 sequence assignments. Supplementary Table 1 contains a complete list of K numbers from all unique wLfag-FL sequences assigned to 146 pathways, as well as their assigned sequence names. Analysis of ANK domain-containing proteins performed on cDNA sequences of wLfag-FL revealed five sequences containing ankyrin domains. This was expected though, as BUSCO analysis revealed only 10.7% of the endosymbiont’s transcriptome was recovered from the four datasets, and that other closely related Wolbachia strains of Supergroup B contain around 50-60 ANK domain proteins. Our identification of 5 ANK domain-containing proteins is consistent with our BUSCO score and average number of ANK domain proteins in Supergroup B strains which predicts that wLfag-FL encodes roughly 50 ANK domain proteins.

Figure 5. Bar graph containing 13 KEGG Orthology pathways having 10 or more sequence assignments from Wolbachia identified in L. fagarae FL-isolates. A full table of sequences assigned to KO identifiers is available in Supplementary Table 1.
The reproductive advantages provided by Wolbachia-host symbiosis are critical aspects of the symbiotic relationship; however, Wolbachia provides another method to increase host fitness by supporting nutritional provisioning. A 5’ portion of the Wolbachia bacterioferritin CDS was present in wLfag-FL, having the closest similarity to bacterioferritin characterized in the Wolbachia endosymbiont of Homalodisca vitripennis (MBR9983968.1). To verify the identity of the partial bacterioferritin transcript, we used ExPASy translate to identify the correct protein sequence and aligned previously characterized bacterioferritin protein sequences, discovered by methods other than bioinformatic analysis, with MUSCLE. We found the highly conserved residues within the ferroxidase center to align between all 8 sequences [Figure 6]. Supplementary Table 2 contains accession numbers of sequences used in the bacterioferritin protein alignment. Furthermore, direct comparisons of the wLfag-FL transcriptome to full and partially sequenced Wolbachia genomes from Diaphorina citri hosts identified protein domains of phytanoyl-CoA dioxygenase and collagen-like proteins associated with metabolic processes in wLfag-FL and other Wolbachia strains, which are seemingly absent in wDcit sequences.

Figure 6. MUSCLE alignment of 8 bacterioferritin sequences. Residues with a 100% identity between all 8 sequences are highlighted. Sequence accessions are available in Supplementary Table 2.
Proteins secreted by a disease vector are of significance on account of their central role within a broad range of processes through which an organism can elicit infection and evoke intercellular communication. Of all sequences identified by KEGG as belonging to the secretion system pathways, two were affiliated with Type I secretion (T1SS), four with Type IV secretion (T4SS), and five with the Sec-pathway where one of the five was identified as the signal recognition particle (SRP) protein encoded by the ffh gene, which was identified as the prokaryotic counterpart of the eukaryotic 54 kDa subunit of SRP (SRP54). The other Sec-pathway components identified were secA, an ATPase, as well as secD, secE, and secF, which are all integral membrane proteins. Gram-negative bacteria T1SS outer membrane protein TolC was identified along with a T1SS system ATPase. T4SS proteins VirB4, VirB6, VirB9, and VirB11 were additionally identified.
DISCUSSION
Wolbachia is a maternally inherited bacterium pervasively found in insect species[38]. The number of studies surrounding Wolbachia has increased due to an expanded understanding of the microorganism’s influence on reproductive systems, fitness, and development traits in its host. In some insects, Wolbachia are known to upregulate egg production in maternal hosts[18,21,39], and increase recombination of the host X chromosome, resulting in proportionately high genetic divergence[40]. Our study presents the first characterization of Wolbachia in L. fagarae and our results from MLST and wsp analysis indicate we have recovered transcripts from a new strain. Therefore, based on prior classification per the established nomenclature system[41], we propose the designation wLfag-FL as the species strain ID of the Wolbachia isolate from wild lime psyllids in the state of Florida, United States.
To identify the strain of Wolbachia, multiple housekeeping genes circulated over the genome were used as a core marker gene set, which is suggested for accurate Wolbachia strain genotyping[42]. The wsp gene sequence was further utilized to depict relations to other Wolbachia strains without full genome assemblies. In both analyses, wLfag-FL consistently grouped with Wolbachia from Supergroup B, which contains other closely related psyllid species known to house disease-causing Liberibacteria. We also found that wLfag-FL is closely related to the Wolbachia resident of B. cockerelli, further supporting that both insects originated from Central America. One other important insect pest of South and Central America is the coffee berry borer Hypothenemus hampei, which is credited for causing losses of more than a half billion USD in the coffee-producing industry[43]. H. hampei origins are traced to tropical biomes of Africa and its Wolbachia endosymbiont grouped with Drosophila melanogaster, another insect of African origin. The placement of other Wolbachia strains into the correct supergroups strengthens the interpretation of our MLST analysis.
Regarding endosymbiotic Wolbachia, one of the most intriguing components for host interaction is the substantial number of genes encoding proteins with ANK domains. As few as five ANK domain-containing proteins were identified in the Wolbachia endosymbiont of Brugia malayi, and as many as 60 were identified in the Wolb-pip strain of Wolbachia from the Culex pipiens group[44]. Annotation of the Wolbachia endosymbiont of the Diaphorina citri genome revealed 54 predicted proteins containing ANK domains[45]. ANK genes in Wolbachia have been proposed as major factors in the endosymbiont’s functional role and unique symbiotic relationship. Functions of ANK-containing proteins acting as binding molecules in Wolbachia are difficult to predict since few have sequence similarity outside the ankyrin domains to other proteins of known functions[46]. Nevertheless, many studies suggest that Wolbachia ankyrin domain proteins are involved in many symbiotic pathways, expanding the understanding of how intertwined Wolbachia-host interaction truly is. Biological processes such as the assembly of cation channels, transcriptional regulation, cell differentiation, apoptosis, cell signaling, and most importantly, secretion systems are all thought to contain some sort of protein embodying the ANK motif[47-49]. Further analysis of ANK domain proteins of wLfag-FL must be conducted to obtain a clearer understanding of their roles in this specific Wolbachia-host symbiosis, as our recovery of only 5 transcripts is not enough to draw extensive conclusions.
By influencing host metabolic processes, Wolbachia enhance their selection process as the reproductive assistance they provide may not benefit all hosts selected for infection. Provided the extensive gene loss in endosymbiotic Wolbachia, it is inferred that these strains are nutritionally dependent upon their hosts. Bacterioferritin, the bacterial homolog of eukaryotic ferritin, has been inferred to help eukaryotic hosts capture excess Fe(2+) to avoid iron toxicity and allow the ferric mineral to have increased bioavailability[50]. The three coordination environments (A, B, C) in the ferroxidase center of bacterioferritin are highly conserved, consisting of four alpha helices (A, B, C, and D). Helix A is characterized by two highly conserved αα’s Glu18, and Tyr25, Helix B by αα’s Glu47, Glu51, and His54, Helix C by αα Glu94, and Helix D by αα’s Asp126, Glu127, Gly129, and His130[51]. All the listed conserved residues were shown to exist in wLfag-FL bacterioferritin, except the residues of Helix D, as the translated protein sequence obtained from wLfag-FL accounts only for the first 95 residues of the Wolbachia bacterioferritin protein sequence.
Other predicted metabolites and transporters have been identified in wLfag-FL, inferring the provision of biological molecules for the metabolism of riboflavin, glutathione, glucose, carbon, pentose phosphates, pyrimidines, purines, fatty acids, and various amino acids. Transporters associated with various substrates including pyruvate, carbohydrates, amino acid catabolism, and inorganic cations were additionally identified[46,52,53]. Analysis of the wLfag-FL transcriptome identified a sequence encoding a phytanoyl-CoA dioxygenase family protein, which catalyzes conversions of alpha-ketoglutarate to succinate, and phytanoyl-CoA to 2-hydroxyphytanoyl-CoA[54]. The generation of succinate by phytanoyl-CoA dioxygenase, a family member of iron(II)-dependent oxygenases, has been suggested to aid in energy production under conditions of nutritional stress[55,56]. It has been proposed that in predicted operons combining 2OGFeDOs of phytanoyl CoA oxygenase with sulfotransferases, there are collagen-like domain proteins thought to act as substrates of the related co-encoded enzymes[57]. A collagen-like protein coding sequence, containing 20 copies of a collagen triple helix repeat identified by Pfam v34.0[58], was found in wLfag-FL and has homology to another collagen-like protein identified in Wolbachia endosymbionts containing the beforementioned phytanoyl-CoA dioxygenase family protein. However, this specific collagen-like protein had no homology to collagen-like proteins identified in wDcit endosymbionts, suggesting collaboration with a hydroxylation system no longer present in the Wolbachia resident of Diaphorina citri. Other protein coding regions of interest involved in various processes were identified in wLfag-FL that had no homology to coding regions within wDcit genome assemblies. These sequences encoded for S-adenosylmethionine uptake transporter, UDP-glucose 6-dehydrogenase, and an IS66 family transposase. S-adenosylmethionine uptake transporter of the DMT-superfamily contributes to the transport of S-adenosylmethionine required by various biological pathways. UDP-glucose 6-dehydrogenase is associated with outer cell surface glycostructures such as lipopolysaccharide biosynthesis. IS66 family transposase is necessary for DNA transposition and may cause structural variations in bacterial plasmids. Further research is necessary to identify other gene products involved with the three mentioned proteins, as around 10% of the transcriptome was recovered. Enrichment for this new Wolbachia strain and deeper sequencing must be conducted to detect other genetic elements, enabling full transcriptome or genome comparisons. As Wolbachia exhibit important roles with an insect’s microbiome, identification of genes necessary to uphold the symbiotic relationship may provide targets for RNAi.
Fundamental pathways used to assess the combativeness of an infection are those containing secreted proteins. KEGG analysis revealed proteins from the T1SS, T4SS, and Sec pathways in wLfag-FL. From the Sec-pathway, SRP ffh was identified and has been shown to work in cohorts with a ribosome-associated chaperone and peptidyl-prolyl isomerase, which determine whether an actively synthesized protein will be inserted into the outer membrane or exported through T2SS[59]. Provided the conservation of the ffh coding region between Wolbachia strains, it may serve as a component in future MLST studies to assist in the further classification of newly sequenced Wolbachia strains. The T4SS system is one of several types of secretion systems utilized by microorganisms for the transport of macromolecules across the cell envelope, and disruption of this pathway, preventing protein oligomerization, may invoke lower infection rates of various bacterial species. Lastly, the identification of T4SS Vir proteins suggests that both operons within the virB-virD4 loci are present in wLfag-FL[60].
Limitations
Recovery of the complete Wolbachia transcriptome from the four cDNA datasets was not successful, as determined by our low BUSCO scores and low percentage of unique transcripts mapped to transcripts of related Wolbachia. We believe this low number of complete transcripts is due to the fragmented nature of the transcript assembly and the inability of BUSCO to identify suitable protein alignments. Our N50 value is relatively low compared to N50s of 1,215 for wDcit dawsonii and 1,236 for wSpic. We attempted to further assemble the transcripts using CAP3, which negligibly increased our N50 to 751; however, this resulted in a lower complete BUSCO score. Figures 3 and 4 show a complete distribution of transcripts throughout whole reference genomes, and not just 15% of the genomes, which would be expected considering our BUSCO score. Short transcripts, around 150-250 bp, still mapped regularly throughout entire reference genomes, suggesting their presence, although complete annotation was not possible through BUSCO or KEGG analysis. This assessment was not surprising as Wolbachia was not the target of initial sequencing efforts and, therefore, sequencing of nucleic acids enriched for microorganisms by a pretreatment DNase and filtration protocol[61] must be performed for a complete assessment of the wLfag-FL transcriptome. Furthermore, long-read sequencing of the genomic DNA will reveal a more complete assessment of the total CDS encoded in the wLfag genome.
In conclusion, The comprehensive screening and classification of Wolbachia in L. fagarae has broadened the host spectrum of Wolbachia identified in members of Hemiptera: Psylloidea. A detailed MLST classification of conserved housekeeping genes is standard practice and an effective method to determine the phylogenetic classification of genetically divergent Wolbachia strains. The newly identified Wolbachia strain in L. fagarae provides conserved biological elements that may be manipulated to influence host vector fitness and fecundity. This study allows for the advancement of biological control applications that do not involve the use of environmentally harmful chemicals, thus focusing on environmentally safer management strategies for pest control while protecting the ecosystem.
DECLARATIONS
Acknowledgments
The authors thank Mrs. Maria Gonzalez, Biological Science Technician, ARS; Dr. Surya Saha, Bioinformatics Advisor; and Dr. Thomson Paris, DOE/USDA, ORISE participant, sample collection. This research was supported in part by an appointment to the Agricultural Research Service (ARS) Research Participation Program administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and the U.S. Department of Agriculture (USDA). ORISE is managed by ORAU under DOE contract number DE-SC0014664. All opinions expressed in this paper are the authors’ and do not necessarily reflect the policies and views of USDA, ARS, DOE, or ORAU/ORISE.
Authors’ contributions
Project design: Hunter WB
Conceptualization: Hunter WB, Stuehler DS Jr.
Methodology: Stuehler DS Jr., Cano LM, Qureshi JA
PCR screening and sequencing: Cano LM, Qureshi JA, Hunter WB
Molecular analysis: Stuehler DS Jr., Cano LM
Writing - original draft preparation: Stuehler DS Jr.
Writing - review and editing; Hunter WB, Cano LM
Figure preparation: Stuehler DS Jr., Hunter WB
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
The data supporting this work is provided in the text. Additional raw data will be made available by the corresponding author upon reasonable request.
Financial support and sponsorship
The financial support for this study was provided to Hunter WB, Qureshi JA, and Cano LM by NIFA CAP Grant 2020-70029-33176 “Therapeutic Molecule Evaluation and Field Delivery Pipeline for Solutions to HLB”, NIFA USDA Citrus Greening award #2015-70016-23028, “Developing an Infrastructure and Product Test Pipeline to Deliver Novel Therapies for Citrus Greening Disease”, and NIFA Hatch project FLA-IRC-006515 “Deploying genome-informed pipelines and integrating transcriptome analyses of pathogen-host interactions”. The Agricultural Research Service (ARS) Research Participation Program administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the US Department of Energy (DOE) and the US Department of Agriculture (USDA), ORISE agreement, 60-6034-1-002. ORISE is managed by ORAU under DOE contract number DE-SC0014664. All opinions expressed in this study are the authors’ and do not necessarily reflect the policies and views of USDA, ARS, DOE, or ORAU/ORISE.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
1. Russell DN, Qureshi JA, Halbert SE, Stansly PA. Host suitability of citrus and Zanthoxylum spp. for Leuronota fagarae burckhardt and Diaphorina citri kuwayama (Hemiptera: Psylloidea). Fla Entomol. 2014;97:1481-92.
2. Burckhardt D. Jumping plant lice (Homoptera: Psylloidea) of the temperate neotropical region. Part 3: calophyidae and triozidae. Zool J Linn Soc. 1988;92:115-91.
3. Chrostek E, Pelz-Stelinski K, Hurst GDD, Hughes GL. Horizontal transmission of intracellular insect symbionts via plants. Front Microbiol. 2017;8:2237.
4. Sintupachee S, Milne JR, Poonchaisri S, Baimai V, Kittayapong P. Closely related Wolbachia strains within the pumpkin arthropod community and the potential for horizontal transmission via the plant. Microb Ecol. 2006;51:294-301.
5. Hoffmann AA, Cooper BS. Describing endosymbiont-host interactions within the parasitism-mutualism continuum. Ecol Evol. 2024;14:e11705.
6. Baumann P. Biology bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu Rev Microbiol. 2005;59:155-89.
7. Benlarbi M, Ready PD. Host-specific Wolbachia strains in widespread populations of Phlebotomus perniciosus and P. papatasi (Diptera: Psychodidae), and prospects for driving genes into these vectors of Leishmania. Bull Entomol Res. 2003;93:383-91.
8. Charlesworth J, Weinert LA, Araujo EV Jr, Welch JJ. Wolbachia, Cardinium and climate: an analysis of global data. Biol Lett. 2019;15:20190273.
9. Hague MTJ, Caldwell CN, Cooper BS. Divergent effects of Wolbachia on host temperature preference. bioRxiv 2020. Available from: http://biorxiv.org/lookup/doi/10.1101/2020.06.11.146977. [Last accessed on 31 Mar 2025]
10. Werren JH, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741-51.
11. Manthey JD, Girón JC, Hruska JP. Impact of host demography and evolutionary history on endosymbiont molecular evolution: a test in carpenter ants (genus Camponotus) and their Blochmannia endosymbionts. Ecol Evol. 2022;12:e9026.
12. Bontemps Z, Paranjape K, Guy L. Host-bacteria interactions: ecological and evolutionary insights from ancient, professional endosymbionts. FEMS Microbiol Rev. 2024;48:fuae021.
13. Kraus S, Monchanin C, Gomez-Moracho T, Lihoreau M. Insect diet. In: Vonk J, Shackelford T, editors. Encyclopedia of animal cognition and behavior. Cham: Springer International Publishing; 2019. p. 1-9.
14. Douglas AE. Microbial nutrient factories in insects on extreme diets. Comptes Rendus Biol. 2019;342:260.
15. Fagen JR, Giongo A, Brown CT, Davis-Richardson AG, Gano KA, Triplett EW. Characterization of the relative abundance of the citrus pathogen Ca. Liberibacter asiaticus in the microbiome of its insect vector, Diaphorina citri, using high throughput 16S rRNA sequencing. Open Microbiol J. 2012;6:29-33.
16. Zug R, Hammerstein P. Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One. 2012;7:e38544.
17. Roldán EL, Stelinski LL, Pelz-Stelinski KS. Reduction of Wolbachia in Diaphorina citri (Hemiptera: Liviidae) increases phytopathogen acquisition and decreases fitness. J Econ Entomol. 2024;117:733-49.
18. Bi J, Wang YF. The effect of the endosymbiont Wolbachia on the behavior of insect hosts. Insect Sci. 2020;27:846-58.
19. Saucereau Y, Valiente Moro C, Dieryckx C, et al. Comprehensive proteome profiling in Aedes albopictus to decipher Wolbachia-arbovirus interference phenomenon. BMC Genomics. 2017;18:635.
20. Yuan LL, Chen X, Zong Q, et al. Quantitative proteomic analyses of molecular mechanisms associated with cytoplasmic incompatibility in Drosophila melanogaster induced by Wolbachia. J Proteome Res. 2015;14:3835-47.
21. Guo Y, Hoffmann AA, Xu XQ, et al. Vertical transmission of Wolbachia is associated with host vitellogenin in Laodelphax striatellus. Front Microbiol. 2018;9:2016.
22. Lefoulon E, Clark T, Borveto F, et al. Pseudoscorpion Wolbachia symbionts: diversity and evidence for a new supergroup S. BMC Microbiol. 2020;20:188.
23. Cicero JM, Brown JK. A stationary tweezer platform for high throughput dissections of minute arthropods and extirpation of their minute organs. MethodsX. 2021;8:101317.
24. Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644-52.
25. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907-15.
26. Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 2021;38:4647-54.
27. Okonechnikov K, Golosova O, Fursov M; UGENE team. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012;28:1166-7.
28. Uday J, Puttaraju HP. Comparative analysis of Wolbachia surface protein in D. melanoagster, A. tabida and B. malayi. Bioinformation. 2012;8:711-5.
29. Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28:1947-51.
30. Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545-51.
31. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27-30.
32. Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784-8.
33. Van Meer MM, Witteveldt J, Stouthamer R. Phylogeny of the arthropod endosymbiont Wolbachia based on the wsp gene. Insect Mol Biol. 1999;8:399-408.
34. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2 - a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189-91.
35. Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11:e0163962.
36. Ankenbrand MJ, Hohlfeld S, Hackl T, Förster F. AliTV - interactive visualization of whole genome comparisons. PeerJ Computer Science. 2017;3:e116.
37. Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402.
38. Correa CC, Ballard JWO. Wolbachia associations with insects: winning or losing against a master manipulator. Front Ecol Evol. 2016;3:153.
39. Stouthamer R, Breeuwer JA, Hurst GD. Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annu Rev Microbiol. 1999;53:71-102.
40. Singh ND. Wolbachia infection associated with increased recombination in drosophila. G3. 2019;9:229-37.
41. Zhou W, Rousset F, O’Neil S. Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc Biol Sci. 1998;265:509-15.
42. Baldo L, Dunning Hotopp JC, Jolley KA, et al. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl Environ Microbiol. 2006;72:7098-110.
43. Chapman EG, Messing RH, Harwood JD. Determining the origin of the coffee berry borer invasion of Hawaii. Ann Entomol Soc Am. 2015;108:585-92.
44. Klasson L, Walker T, Sebaihia M, et al. Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol Biol Evol. 2008;25:1877-87.
45. Saha S, Hunter WB, Reese J, et al. Survey of endosymbionts in the Diaphorina citri metagenome and assembly of a Wolbachia wDi draft genome. PLoS One. 2012;7:e50067.
46. Wu M, Sun LV, Vamathevan J, et al. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004;2:E69.
47. Al-Khodor S, Price CT, Kalia A, Abu Kwaik Y. Functional diversity of ankyrin repeats in microbial proteins. Trends Microbiol. 2010;18:132-9.
48. Hryniewicz-Jankowska A, Czogalla A, Bok E, Sikorsk AF. Ankyrins, multifunctional proteins involved in many cellular pathways. Folia Histochem Cytobiol. 2002;40:239-49.
49. Voronin DA, Kiseleva EV. [Functional role of proteins containing ankyrin repeats]. Tsitologiia. 2007;49:989-99.
50. Kremer N, Voronin D, Charif D, Mavingui P, Mollereau B, Vavre F. Wolbachia interferes with ferritin expression and iron metabolism in insects. PLoS Pathog. 2009;5:e1000630.
51. Ebrahimi K, Hagedoorn PL, Hagen WR. Unity in the biochemistry of the iron-storage proteins ferritin and bacterioferritin. Chem Rev. 2015;115:295-326.
52. Foster J, Ganatra M, Kamal I, et al. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 2005;3:e121.
53. Nikoh N, Hosokawa T, Moriyama M, Oshima K, Hattori M, Fukatsu T. Evolutionary origin of insect-Wolbachia nutritional mutualism. Proc Natl Acad Sci U S A. 2014;111:10257-62.
54. Baldridge GD, Markowski TW, Witthuhn BA, Higgins L, Baldridge AS, Fallon AM. The Wolbachia WO bacteriophage proteome in the Aedes albopictus C/wStr1 cell line: evidence for lytic activity? In Vitro Cell Dev Biol Anim. 2016;52:77-88.
55. Gan HM, Hudson AO, Rahman AY, Chan KG, Savka MA. Comparative genomic analysis of six bacteria belonging to the genus Novosphingobium: insights into marine adaptation, cell-cell signaling and bioremediation. BMC Genomics. 2013;14:431.
56. Sullivan MB, Huang KH, Ignacio-Espinoza JC, et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 2010;12:3035-56.
57. Iyer LM, Abhiman S, de Souza RF, Aravind L. Origin and evolution of peptide-modifying dioxygenases and identification of the wybutosine hydroxylase/hydroperoxidase. Nucleic Acids Res. 2010;38:5261-79.
58. Mistry J, Chuguransky S, Williams L, et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021;49:D412-9.
59. Beck K, Wu LF, Brunner J, Müller M. Discrimination between SRP- and SecA/SecB-dependent substrates involves selective recognition of nascent chains by SRP and trigger factor. EMBO J. 2000;19:134-43.
60. Rancès E, Voronin D, Tran-Van V, Mavingui P. Genetic and functional characterization of the type IV secretion system in Wolbachia. J Bacteriol. 2008;190:5020-30.
Cite This Article
 , ... Liliana M. Cano
, ... Liliana M. CanoHow to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].