


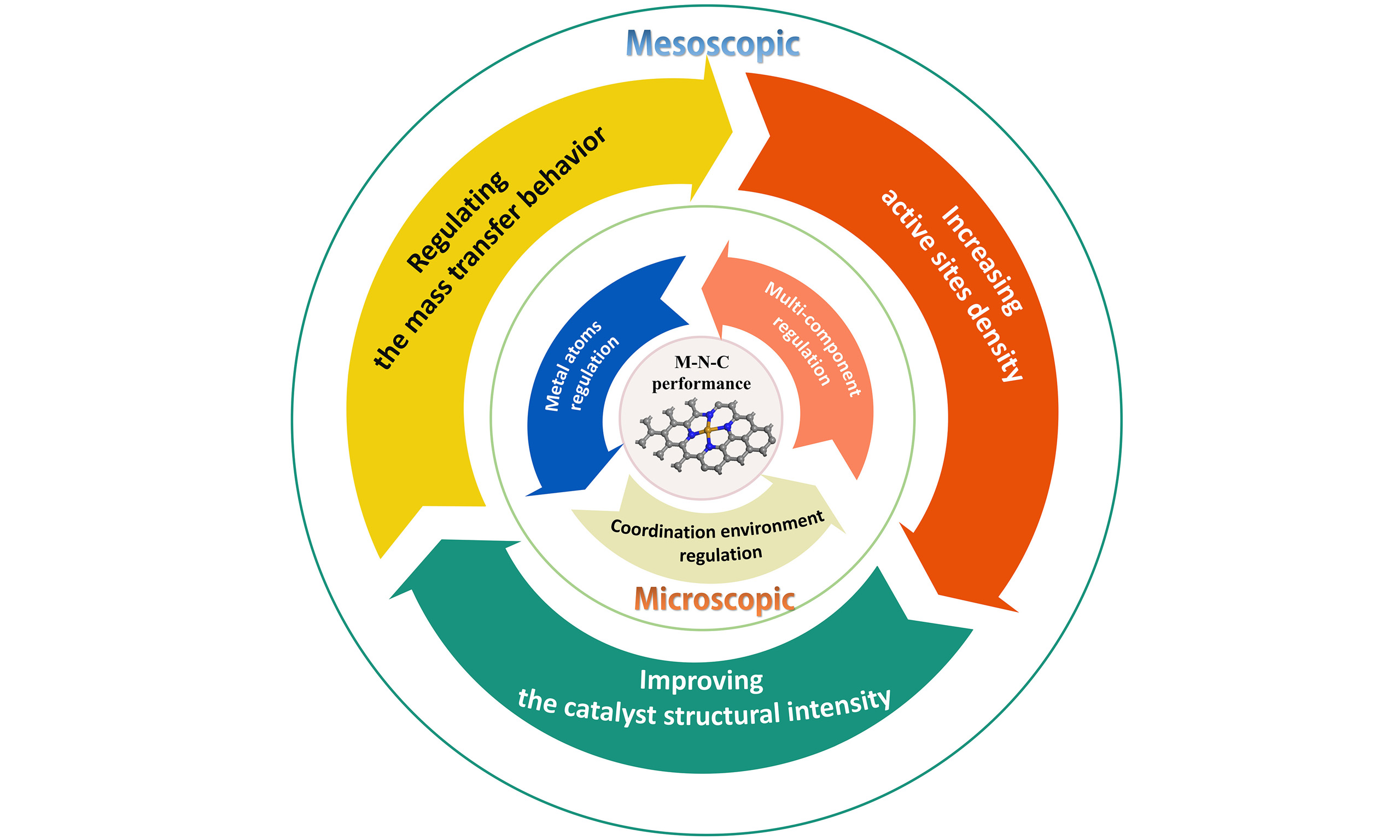
Multiscale designing principle of M-N-C towards high performance PEMFC
 0
0 Abstract
Among the reported non-precious-metal catalysts, metal-nitrogen-carbon (M-N-C) catalysts have emerged as a research cornerstone in the field of electrocatalysis, showcasing unparalleled activity in oxygen reduction reactions that rivals or even exceeds that of commercial Pt catalysts. Despite boasting high atom utilization and adjustable effective activity centers, M-N-C catalysts suffer from inadequate long-term stability under high-pressure and harshly acidic conditions within proton exchange membrane fuel cells (PEMFCs). This drawback poses a significant challenge that critically limits their potential for widespread applications. From this perspective, we commence by delineating the pivotal strategies to augment the performance of M-N-C catalysts at the microscopic level, including the tuning of the intrinsic activity of individual active sites and the manipulation of their quantity. Furthermore, we delve into the benefits derived from the synergistic effects unleashed by the incorporation of multi-component active sites. At the mesoscopic level, this perspective engages with the design principles aimed at enhancing the activity and stability of M-N-C catalysts within the intricate three-phase boundary of PEMFCs. Ultimately, we prospect the opportunities and challenges facing the future evolution of M-N-C catalysts, with the aim of offering comprehensive guidance for the design and advancement of highly stable M-N-C catalysts tailored for PEMFC applications.
Keywords
INTRODUCTION
Hydrogen energy is renowned for its substantial calorific value, varied sources, ample reserves, and distinctive benefits for large-scale and extended-duration storage, earning it the distinction of being the ultimate energy source for the 21st century[1,2]. Among the most notable terminals for hydrogen energy application, the proton exchange membrane fuel cell (PEMFC), with high energy conversion efficiency and emission of non-polluting byproducts, has gained wide attention in the pursuit of efficient harnessing of clean energy, reduction of carbon emissions, and the mitigation of environmental contamination[3-5]. However, the oxygen reduction reaction (ORR) occurring at the cathode remains a persistent bottleneck in the advancement of PEMFC. As a multistep, complex reaction including four proton-coupled electron transfers (PCETs), the kinetic process is quite sluggish, leading to the requirement of massive use of Platinum-group metal (PGM) catalysts for accelerating the reaction, which significantly increases the cost of the entire cell system[6-8]. PGMs are well known for their exorbitant prices and limited global availability; thus, the current development of PGM-free catalysts with high activity and stability to bridge the gap with standard commercial PGM catalysts has become a roadmap to be reckoned with[9,10].
Since the concept of single-atom catalysts (SACs) was proposed in 2011, these low-cost and feedstock-source-rich catalysts have unlocked a multitude of new applications. Based on the principle of maximizing the accessibility of active metal atoms by minimizing the size of the metal ensemble, SACs have demonstrated remarkable prominence as a beneficial solution for cost reduction in PEMFC and ORR[11-13]. N-doped carbon-based transition metal (Fe, Co, and Mn) SACs, also known as metal-nitrogen-carbon
For SACs such as M-N-C, it is inherently presumed that the enhancement of catalytic performance can be achieved through the precise manipulation of the attributes of the individual atomic sites at the microscopic level, as well as through the strategic design of the catalyst’s structure at the mesoscopic scale. In the development of the last decade, the activity of the most promising Fe-N-C catalysts has been gradually refreshed, approached or even exceeded that of Pt/C catalysts[19,20]. This Perspective will focus on recent PEMFC-oriented M-N-C catalyst activity and stability pivotal issues and advancements, aiming to discuss the concepts of intrinsic activity enhancement of the catalyst active centers and the optimization of the overall catalyst performance across both microscopic and mesoscopic dimensions. Typical deactivation mechanisms of M-N-C catalysts and corresponding inhibition strategies are also covered to facilitate the design of next-generation M-N-C electrocatalysts to meet practical requirements.
OPTIMIZING INTRINSIC CATALYTIC PERFORMANCE IN MICROSCOPIC
Metal atoms regulation
Typically, ORR involves a four-electron (4e-) pathway and a two-electron (2e-) pathway. For the 4e- pathway, the elementary steps are as follows:
O2 + * + H2O + e- → OOH* + OH-
OOH* + e- → O* + OH-
O* + H2O + e- → OH* + OH-
OH* + e- → * + OH-
While the 4e- pathway can be achieved as follows:
O2 + H2O + * + e- → OOH* + OH-
OOH* + H2O + e- → * + H2O
Thus, the reactivity of ORR undergoing the 4e- or 2e- pathway relies on the adsorption mode and reaction barrier of adsorption intermediates on the catalyst surface[21]. The 4e- pathway reaction of ORR involves various key intermediates such as *OOH, *O, and *OH [Figure 1A][22], and according to the well-known volcano plot constructed by Nørskov et al. [Figure 1B][23], the binding strength of the above intermediates is highly related to ORR activity and selectivity of the above pathways[23,24]. Moreover, this volcano plot indicates that Pt has the highest intrinsic ORR activity while transition metals such as Fe, Co, Mn, Ni and Zn exhibit low ORR activity. This limitation hinders efforts to reduce the cost of ORR catalysts. Thus, tuning the binding energy of key intermediates during ORR on transition metals is of critical importance for the large-scale commercialization of ORR catalysts. In M-N-C systems, M bonds with x (x higher than 4) N atoms, which connect to C framework with C-N bond, to form MNx moieties which are widely considered as the active sites of ORR [Figure 1C][25]. Recent studies show that modifying electronic structure of SACs is critical for changing the binding energy of key ORR intermediates and thus significantly influences the ORR catalytic performance. Most of the well-known electronic structure modification strategies involve the microstructure-manipulation of MNx moieties. For a long time, Fe SACs have been considered excellent

Figure 1. (A) Schematics of ORR 2e- pathway and 4e- pathway with the corresponding intermediates[22]; (B) Trends in oxygen reduction activity plotted as a function of the oxygen binding energy[23]; (C) Schematic model of Fe-N-C. Fe (orange), N (blue), O (red), and C (gray)[25]; (D) Free energy diagrams of 2e- ORR on the SACs at U = 0.7 V versus RHE[28]; (E) Schematic illustration for the formation of Co SAs/N-C through pyrolysis of Zn/Co MOFs[29]; (F) Thermodynamic-activity relation trends for the 2e- pathway for H2O2 product of different M-N-C[30]; (G-H) In situ TEM images in synthetic process of Fe SAs/S-NC (Fe3O4@SiO2/CdS@PDA)[32]; (I) Aberration-corrected HAADF-STEM images corresponding to the selected area in (H) of Fe SAs/S-NC[32]. ORR: Oxygen reduction reaction; M-N-C: metal-nitrogen-carbon; SACs: single-atom catalysts; Fe SAs/S-NC: Fe single atoms arranged on S, N co-doped carbon skeleton; RHE: reversible hydrogen electrode; MOFs: metal-organic frameworks; HAADF-STEM: high-angle annular dark-field scanning transmission electron microscopy.
Coordination environment regulation
The appropriate selection of the center metal and coordination atoms and the tuning of the local coordination environments are crucial for improving the intrinsic activity of the catalysts. By tailoring the geometric configuration of N species to adjust the coordination environment around metal atoms, the electron density and d-orbital occupancy at the active center can be finely controlled, ultimately refining the binding energy of critical intermediates in the ORR process. Concurrently, manipulating the spin state of the central metal atoms yields a wealth of unpaired d-orbital electrons, thereby enhancing the M-O bonding interaction between the active sites and the pivotal ORR intermediates. In general, the interaction between key intermediates and MNx moieties would be well manipulated by tuning the electronic properties of active sites with specific microstructure, making M-N-C SACs promising materials for high ORR electrocatalytic performance. Adjusting the coordinate atom is one of the commonly applied methods regulating the coordination environment for higher ORR performance. The introduction of O and S into MNx moieties would modulate the binding energy of key ORR intermediates which eventually regulate the reaction pathway. Sun et al. designed a high ORR activity O-doped O-FeN4C-O catalyst and found that the introduction of O would enhance the energy of Fe 3d orbital, which leads to a strong binding energy between the Fe central atom site and OH[27]. This study demonstrates that the presence of O in FeN4C catalysts tailors the electronic structure of the MNx moieties and optimizes the ORR performance. A recent study also demonstrates that the adsorption of O near Co-N4-C moieties would modulate the electron density of the moieties and enhance the adsorption energy of OOH on Co-active sites[31]. An optimal OOH adsorption energy is obtained with the adsorption of two O near Co-N4 moieties, which leads to high H2O2 selectivity. Tian et al. employed a thermal printing strategy to enable individual Fe3O4 nanoparticles to release metal atoms at elevated temperatures, which were subsequently captured In situ by nitrogen and sulfur defects in the carbon layer to generate Fe single atom sites [Figure 1G-I][32]. The obtained Fe single atoms arranged on S, N co-doped carbon skeleton (Fe SAs/S-NC) exhibited excellent oxygen reduction reactivity; besides, the metal loading could be effectively regulated by controlling the size of the nanoparticles at the interface. A sulfur-doped Mn-N-C catalyst is also proposed for higher ORR activity in acid environment rather than S-free Mn-N-C catalyst, showing promising way regulating the performance of M-N-C catalysts with adjacent coordinative dopants strategy[33].
Besides tailoring the coordination species, modulating the coordination structure also plays a significant role in coordination environment regulating strategy. Recently, Hu et al. studied Fe-N-C catalysts with 13 different N-coordinated configurations to reveal the relationship between the coordination structure and the ORR performance with density functional theory (DFT) calculation[34]. They demonstrate that Fe-N-C with the coordination structure of pyrrolic N shows higher ORR activity rather than the pyridinic coordination structure. Moreover, the pyrrolic FeN4C catalyst has the highest ORR activity among all pyrrolic ones, which indicates an increasing activity relating to an increasing coordinating N content. For Co-N-C, the type of the coordination structure also influences the ORR selectivity. A recent study shows that the pyrrole-type Co-N-C has higher 2e- ORR selectivity while the pyridine-type Co-N-C prefers 4e- ORR[35]. The origin of this difference mainly contributes to the different adsorption energies between CoN4 and OOH intermediates of varying coordination structures. The above studies provide fundamental insight into the rational design strategy of coordination environment.
Multi-component regulation
The single-metal center in M-N-C presents a challenge in overcoming the scaling relationship inherent in the ORR process. Incorporating additional components allows for a transcendence of this constraint, thereby surmounting the impediment to enhanced catalytic efficiency associated with single-site catalysts[36-38]. In recent years, the combination of single atoms with other atoms, clusters, and even nanoparticles of multiscale metals to form hybrid catalysts has spearheaded a new wave of synergistic catalysis. The heterogeneous electrocatalysts reported in 2018 containing both Pt-Co alloy particles, Co particles, and Co-Nx-Cy sites were among the leading ones, demonstrating in the time 12.36 A mgPt-1@0.9 V ORR breakthrough mass activity, which greatly stimulated the interest of later developers [Figure 2A-C][39]. In the hybrid system, M-N-C is uniquely advantageous as carbon support rich in both ORR active centers and N species, and when coupled with Pt and Pt-alloyed nanoparticles, unexpected activity and durability enhancements were obtained while achieving catalyst cost control. Guo et al. presented a closely packed hybrid electrocatalyst containing Pt-based alloy nanocrystals and dense isolated Ni sites, which ensured the relay reduction of the two electrons at Ni single sites, and the subsequent two electron transfer on the neighboring Pt-based nanocrystals [Figure 2D][40]. The electronic interaction between single atoms and metal clusters represents a significant potential for improving SACs by optimizing the electron transfer process. As detailed by Wan et al., the incorporation of Fe clusters into Fe-N4 sites has resulted in a notable enhancement of both activity and stability with approximately a 60% increase in each aspect[41]. Due to unimpeded electron transfer pathways and short interaction distances, Fe clusters and Fe-N4 sites have established strong electronic interactions. Specifically, the Fe clusters optimized the adsorption strength of redox intermediates on Fe-N4 sites and shortened the bond amplitude of Fe-N4 through incoherent vibrations. Furthermore, the employment of dual-atom catalysts, achieved through the integration of a “second atom”, can enhance catalytic performance by manipulating atomic density, spatial configuration, and positioning, as well as electron distribution, via a spectrum of interactions such as synergistic effects, lattice strain enhancement, and electronic modulation[42-44]. The (Fe, Co)/N-C catalyst developed by Wang et al. has been demonstrated to be more favorable for O-O bond breaking in the ORR process and has exhibited superior performance in practical applications in hydrogen-oxygen and hydrogen-air fuel cells

Figure 2. (A) Schematics of heterogeneous electrocatalysts showing coexistence of Pt-Co NPs, Co@graphene, and Co-N4-C PGM-free active sites (named LP@PF)[39]; (B) HAADF-STEM image of Pt-Co NPs in the heterogeneous electrocatalysts[39]; (C) HAADF-STEM image of PGM-free support containing atomically dispersed Co (circled in red) and trace Pt (circled in blue)[39]; (D) Schematic illustration for Preparing closely packed Pt-based alloy nanocrystals and dense isolated Ni sites hybrid (Pt1.5Ni1-x/Ni-N-C)[40]; (E-G) HAADF-STEM images of Fe-Co dual sites embedded on N-doped porous carbon [(Fe, Co)/N-C][45]; (H) Schematic for well-dispersed Ta-TiOxnanoparticles on the carbon substrate as scavengers[49]; (I) Decay kinetics of the OH-DMPO·as a function of time with and without the addition of Ta-TiOx/KB (the supported Ta-TiOx nanoparticles on a Ketjenblack substrate)[49]; (J) Cell voltage and power density polarization (K) for Fe-N-C with and without the addition of Ta-TiOx/KB before and after the accelerated durability test (ADT)[49]. PGM: Platinum-group metal; NPs: nanoparticles; HAADF-STEM: high-angle annular dark-field scanning transmission electron microscopy.
The specificity of the catalyst towards the desired reduction pathway is of paramount importance. In addition to the direct 4e- pathway, hydrogen peroxide (H2O2) generated in the incomplete 2e- pathway tends to attack the active center of the M-N-C single metal site, resulting in the rapid generation of reactive oxygen species (ROS)[47]. Specifically, the Fe2+ and oxygen in the catalyst react in an acidic environment to form H2O2 to initiate the Fenton reaction. Subsequently, in the presence of Fe2+, H2O2 can be further decomposed to produce ROS such as hydroxyl radicals (-OH). The highly oxidizing nature of H2O2 and ROS not only leads to direct oxidation of the metallic active sites and the formation of oxygen-containing functional groups, which markedly diminishes the turnover frequency (TOF) of the catalyst, but also engages in interactions that alter the chemical state of the active centers, resulting in the reduction of the activity of the catalyst or even its inactivation. At the same time, ROS and H2O2 oxidatively corrode the carbon supports leading to structural collapse and dementalization, accelerating the degradation of proton membranes and ionomers, and generating a huge mass-transfer resistance, resulting in a serious degradation of the PEMFC performance. Especially for Fe2+-containing catalysts, the Fenton reaction
OPTIMIZING OVERALL CATALYTIC PERFORMANCE IN MESOSCOPIC
Increasing active site density
As the number of single-atom active sites increases to a certain threshold, the diminished interatomic spacing gives rise to an electronic perturbation effect. This effect may alter the electron distribution around the metal atoms, instigating a process of charge redistribution, which emerges as a pivotal factor influencing the intrinsic catalytic activity. With an optimally adjusted interatomic spacing, the synergistic interaction between adjacent single-atom active centers can yield an optimized electronic structure. This results in a more substantial mass-specific activity and an ORR performance that approaches the kinetic region characteristic of Pt-based catalysts. A study has explored the kinetic behavior of single active centers and the site-spacing effect of adjacent single metal atoms in Fe-N4. When the distance is less than 1.2 nm, the strong interactions between adjacent Fe-N4 sites change the electronic structure, thereby improving the intrinsic activity of the ORR. Until the distance approaches 0.7 nm or decreases further, a slight decrease in ORR activity is observed[51]. Therefore, the design and optimization of M-N-C synthesis based on site-spacing interactions and the development of M-N-C catalysts with controllable single-atom densities or site spacings are effective approaches to further improve the ORR performance.
Conventional M-N-C synthesis typically involves high-temperature (> 900 °C) pyrolysis, which predisposes the system to particle sintering as the metal precursor loading increases. This phenomenon inherently constrains the potential for enhancing the density of active sites[52]. Over the past decade, researchers have introduced a multitude of optimization strategies to forestall metal agglomeration during the synthesis of M-N-C materials. These strategies encompass complexation protection, impregnation adsorption, the use of sacrificial templates, and the spatially restricted domain approach, collectively surmounting the limitations associated with high metal loading in M-N-C[53-56]. In recent years, the low-temperature reduction of M-N-C has also been developed. Yin et al. have successfully realized the low-temperature synthesis of various high-density M-N-C materials, predominantly Fe-N-C (8.3 wt%), which exhibit superior dispersion and PEMFC performance. Their findings suggest that the structural evolution of Fe-N4 and the tuning of its site density can be effectively achieved through precise control of the synthesis temperature and processing time. This level of control allows for direct manipulation of the catalytic properties of Fe-N-C[57]. With more advanced synthesis processes, such as the tuning of the annealing environment, it was also successfully demonstrated as a method for the preparation of ultra-high single-atom loadings. For example, the negative pressure calcination treatment was able to accelerate the removal of anions from the metal feedstock and promote the bonding of the metal to the nitrogen-deficient sites, resulting in the formation of dense nitrogen-coordinated metal sites (7.3-44.8 wt.%) [Figure 3A-B][58]. These rational preparation methods pave a new way to prepare single-atom M-N-C ORR catalysts, incrementally overcoming the challenges associated with the synthesis of high-density M-N-C materials.

Figure 3. (A) Schematic illustration for preparing ultra-high-loading (UHL, higher than 10 wt%) SACs[58]; (B) The metal content in the as-prepared UHL catalysts[58]; (C) Possible structural evolution pathway from an FeN4C12 S1 site to an FeN4C10 S2 site. The grey, blue, white and orange balls represent C, N, H and Fe atoms, respectively. Ef denotes free energy. Blue arrows represent possible structural evolution pathways[19]; (D) Mechanism illustration of the formation of Co single metal sites on three-dimensional graphene frameworks (Co SAs/3D GFs)[68]; (E) Schematic illustration of Acid-alkaline interaction of catalyst-electrolyte interface, i.e., Inactivated TPB: protonation of pyridine N inactivates the active site in mesopores or macropores (> 2.0 nm). Active TPB: Near-surface micropores of 0.8~2.0 nm which are not in direct contact with Nafion. Micropores smaller than 0.8 nm cannot form TPB due to extremely slow mass transfer[70]. SACs: Single-atom catalysts; TPB: triple-phase boundary.
Improving the catalyst structural intensity
Graphitic carbon supports with high symmetry (D6h) and stable π-conjugated electronic networks are inherently fully inert and stable, and thus various strategies to disrupt the π-conjugated electronic network off-domain are necessary to trigger the catalytic activity of carbon-based materials[59]. For SACs with d2h and then diatomic c2v, the ORR activity is elevated with decreasing basal plane symmetry compared to d6h of pristine graphene[54]. However, destabilizing the π-electron network of graphitic carbon implies a sacrifice of the catalyst’s structural stability, and conventional graphitization treatments usually lead to the loss of active sites. Unlike the corrosion of carbon support catalyzed by Pt particles in Pt catalysts, M-N-C electrocatalysts are usually faced with the dissolution of metal sites in acidic and oxidizing environments. The Fenton reaction of the striped metal will exacerbate the oxidative damage to the carbon support, which further results in a vicious cycle of metal stripping-catalyst structure collapse, i.e., a coupling between the stripped metal and carbon corrosion[60]. For example, Liu et al. demonstrated that up to 75% of the Fe-N-C active sites were deactivated by Fe dementalization leading to voltage loss, which subsequently increased the carbon corrosive material in the catalyst layer by a factor of five, reduced the proton transport kinetics by a factor of four, and lowered the rate of the ORR by a factor of three as the main degradation mechanism[61].
Generally, adjusting the pyrolysis temperature represents a sound strategy for designing oxidation-resistant carbon supports, thereby effectively modulating the degree of graphitization within the carbon matrix. It has been demonstrated that the carbon support undergoes partial oxidation during the ORR process, resulting in alterations to the π-electron delocalization properties of the carbon basal plane, which attenuates the binding of oxygen to the Fe-N4 fraction. Hence, beyond the conventional approaches of increasing the graphitization degree of carbon and reducing the density of exposed carbon edges to enhance durability, it is imperative to focus on the bonding strength of the catalyst itself. Strategies to bolster the M-N bond strength and reinforce the interactions between the active center metal and the carbon substrate are essential to prevent the degradation of the catalyst's bonding integrity following the formation of surface CO species. By converting part of the pyrrole N coordinated to the central atom into pyridine N and completing the transition from the FeN4C12 site to the less active but more stable FeN4C10, the energy barrier of the protonation step is elevated, which in turn reduces the possibility of the subsequent occurrence of the Fe-N chemical bond breakage [Figure 3C][19]. The moderate binding energy and the balance between activity and stability are the foremost priorities in the design of all M-N-C electrocatalysts.
Regulating the mass transfer behavior
Facilitate mass transfer
Catalysts regularly exhibit quite different behaviors in aqueous phase electrode measurement environments than in PEMFC, mainly due to the great diversity of reaction interfaces, with the former ORR occurring at the solid-liquid interface, while the latter involves a complex three-phase boundary where the catalyst, the electrolyte, and the oxygen are in contact[62,63]. In actual PEMFC applications, to meet the reactivity demands necessary for the reaction to occur, a higher dosage of M-N-C catalyst is typically needed to ensure effective catalysis. However, this increase in catalyst loading leads to pronounced mass transfer issues[64]. Under operational conditions, the substantial accumulation of water at the cathode of the PEMFC imposes a burden of water flooding on the pores of the M-N-C catalyst. Specifically, this phenomenon hinders the transmission of gaseous reactants to the reaction sites and reduces the active area of the catalyst due to water coverage, which leads to a marked increase in activation overpotential and concentration overpotential within the PEMFC[65]. To address the stringent requirements for rapid mass transfer within gas diffusion electrodes (GDEs), catalysts featuring hierarchical pore structures have been swiftly advanced. These catalysts, with their well-designed mass transfer channels, not only enhance the accessibility of active sites to reactive molecules but also mitigate the issue of water flooding in the micropores of the carbon supports, thereby reducing mass transfer resistance[66,67].
Zhou et al. obtained dense single metal sites (Co, Fe, Ni, etc.) on three-dimensional (3D) graphene frameworks (GFs) by negative pressure pyrolysis, considerably enhancing the accessibility of these metal sites [Figure 3D][68]. The differential pressure between the inside and outside of the metal-organic frameworks (MOFs) induces the cleavage of the derived carbon layer and the enlargement of the mesopores, resulting in a 3D GF with enhanced mesopore and external surface area, which greatly facilitates the mass transfer and utilization of the metal sites. In addition, the hierarchical pore structure can further enhance the utilization of single metal active sites while constructing a maximum three-phase interface. Liu et al. have demonstrated that
Advances in electrochemical molecular probing tools have made it possible to resolve interfacial information in micropores (< 2 nm) and mesopores (2 nm to 50 nm) at higher spatial resolution, and Wang et al. demonstrated that more than 70% of the ORR activity in the PEMFC originated from micropores with sizes ranging from 0.8 nm to 2.0 nm in the Fe/N/C catalyst layer, independent of the microporous surface fraction[70]. More importantly, the catalyst-electrolyte interactions also deserve attention, as Wang et al. reported that the active sites in mesopores or macropores can be deactivated by acid-base interactions between pyridinium N and Nafion in the catalysts, which leads to the deactivation of the three-phase interface [Figure 3E][70]. Therefore, the accurate identification of active and inactive three-phase interfaces also provides a new design principle for M-N-C performance enhancement in PEMFC.
DISCUSSION AND PERSPECTIVES
M-N-C, as a representative SAC widely used in ORR, boasts distinctive advantages in the contemporary evolution of catalysts that strive for high activity, stability, TOF, and reduced reliance on noble or

Figure 4. Schematic illustration for the design principles of M-N-C catalysts at the microscopic and mesoscopic. M-N-C: Metal-nitrogen-carbon.
To break the limitation of scaling relationships, researchers introduce multi-components including metal atoms, clusters, and nanoparticles into M-N-C to constitute a composite system. Through electronic interactions, this strategy can regulate the adsorption and desorption behaviors of the intermediate species and the O-O breaking process more significantly, thus further enhancing the catalytic activity and stability. In addition, the introduction of functional modulators, such as oxides, shows significant effects on scavenging reactive oxygen radicals, reducing the Fenton reaction of reactive metals, and inhibiting the corrosion of carbon supports. In terms of catalyst structural strength, optimization of carbon supports and enhancement of metal-support interactions are also cutting-edge ideas to further improve the activity and stability of PEMFC.
From a more macroscopic point of view, when the actual system comes from the rotating disk electrode (RDE) to the PEMFC level, the basic step of ORR occurs at the electrode-liquid interface, and the mass transfer behavior at the three-phase interface undoubtedly becomes the key issue. The untimely transfer of the carbon support to the liquid and the surface aggregation of oxygen-containing functional groups will enhance the hydrophilicity of the interface, leading to the catalyst flooding phenomenon. A catalyst design that promotes mass transfer can make full use of the active sites and effectively delay the decay of the material due to water flooding, while the rational design of the porous structure and hydrophobic channels is an important prerequisite for promoting mass transfer and constructing a maximum three-phase interface. With the deepening of research, M-N-C materials are expected to perform a greater role in the field of PEMFC, and the opportunities and challenges also put forward higher requirements for the research of M-N-C catalysts. Here, we provide an outlook on future research.
The importance of M-N-C as an ideal mechanism-recognized configuration cannot be overstated. An
Various strategies, including electronic structure modulation and ligand atom optimization, have been broadly implemented to boost the activity of M-N-C catalysts. Nevertheless, M-N-C catalysts continue to grapple with substantial challenges regarding their structural stability and durability under acidic and highly oxidizing conditions compared to Pt catalysts. The impact of the electronic structure of metal atoms on safeguarding active sites and augmenting the structural stability of catalysts through the establishment of robust electronic coupling between the metal and the support is a well-established research area. Strategies such as the utilization of graphitized carbon supports, the encapsulation of surface nitrogen atoms, and the modification of the carbon supports have a firm theoretical foundation and necessitate further detailed investigation. Adjusting the M-N bond to stabilize the metal center and inhibiting the dementalization process is also of profound significance in enhancing the stability of the active sites of M-N-C catalysts. In addition to carbon supports, other supports such as metal oxides, nitrides, carbides, etc., may be promising candidates for single-atom electronic structure tuning to enhance the stability of metal sites due to their complex electronic structures and abundant defect states. In addition, to address the problem of reactive oxygen attack, the development of active center-scavenger composite systems that can effectively avoid free radical attack and reduce the risk of support oxidation is the key to enhancing the performance of M-N-C catalysts in ORR and PEMFC. Advancements in these research avenues will undoubtedly herald novel breakthroughs and opportunities for the enhancement of M-N-C catalyst performance.
DECLARATIONS
Authors’ contributions
Drafted the manuscript: Wu, Y.; Zhou, H.
Participated in the writing, revision and literature discussion: Guo, W.; Liu, Y.
Finalized the manuscript: Guo, W.; Zhou, H.; Wu, Y.
Availability of data and materials
Not applicable.
Financial support and sponsorship
The authors acknowledge funding from China Ministry of Science and Technology (2021YFA1600800), the National Natural Science Foundation of China (22422506, 22201271, 92261105, and 22221003), the Anhui Provincial Natural Science Foundation (2108085UD06, 2208085UD04), the Anhui Provincial Key Research and Development Project (2023z04020010, 2022a05020053), USTC Research Funds of the Double First-Class Initiative (YD2060002029, YD2060006005), and the Joint Funds from Hefei National Synchrotron Radiation Laboratory (KY2060000180 and KY2060000195).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Sartbaeva, A.; Kuznetsov, V. L.; Wells, S. A.; Edwards, P. P. Hydrogen nexus in a sustainable energy future. Energy. Environ. Sci. 2008, 1, 79.
2. Banham, D.; Kishimoto, T.; Zhou, Y.; et al. Critical advancements in achieving high power and stable nonprecious metal catalyst-based MEAs for real-world proton exchange membrane fuel cell applications. Sci. Adv. 2018, 4, eaar7180.
3. Debe, M. K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43-51.
5. Eberle, U.; Müller, B.; von, H. R. Fuel cell electric vehicles and hydrogen infrastructure: status 2012. Energy. Environ. Sci. 2012, 5, 8780.
6. Katsounaros, I.; Schneider, W. B.; Meier, J. C.; et al. Hydrogen peroxide electrochemistry on platinum: towards understanding the oxygen reduction reaction mechanism. Phys. Chem. Chem. Phys. 2012, 14, 7384-91.
7. Gasteiger, H. A.; Kocha, S. S.; Sompalli, B.; Wagner, F. T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B. Environ. 2005, 56, 9-35.
8. Gasteiger, H.; Panels, J.; Yan, S. Dependence of PEM fuel cell performance on catalyst loading. J. Power. Sources. 2004, 127, 162-71.
9. Brouzgou, A.; Song, S.; Tsiakaras, P. Low and non-platinum electrocatalysts for PEMFCs: current status, challenges and prospects. Appl. Catal. B. Environ. 2012, 127, 371-88.
10. Liu, M.; Zhao, Z.; Duan, X.; Huang, Y. Nanoscale structure design for high-performance Pt-based ORR catalysts. Adv. Mater. 2019, 31, e1802234.
11. Yang, X. F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740-8.
12. Qiao, B.; Wang, A.; Yang, X.; et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634-41.
13. He, Y.; Liu, S.; Priest, C.; Shi, Q.; Wu, G. Atomically dispersed metal-nitrogen-carbon catalysts for fuel cells: advances in catalyst design, electrode performance, and durability improvement. Chem. Soc. Rev. 2020, 49, 3484-524.
14. Lefèvre, M.; Proietti, E.; Jaouen, F.; Dodelet, J. P. Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. Science 2009, 324, 71-4.
15. Xie, X.; He, C.; Li, B.; et al. Performance enhancement and degradation mechanism identification of a single-atom Co-N-C catalyst for proton exchange membrane fuel cells. Nat. Catal. 2020, 3, 1044-54.
16. Wan, X.; Liu, X.; Li, Y.; et al. Fe-N-C electrocatalyst with dense active sites and efficient mass transport for high-performance proton exchange membrane fuel cells. Nat. Catal. 2019, 2, 259-68.
17. Li, J.; Chen, M.; Cullen, D. A.; et al. Atomically dispersed manganese catalysts for oxygen reduction in proton-exchange membrane fuel cells. Nat. Catal. 2018, 1, 935-45.
18. The United States Department of Energy. Hydrogen and fuel cell technologies office multi-year research, development, and demonstration plan. Available from: https://www.energy.gov/sites/default/files/2024-05/hfto-mypp-2024.pdf. [Last accessed on 11 Mar 2025].
19. Liu, S.; Li, C.; Zachman, M. J.; et al. Atomically dispersed iron sites with a nitrogen-carbon coating as highly active and durable oxygen reduction catalysts for fuel cells. Nat. Energy. 2022, 7, 652-63.
20. Miao, Z.; Wang, X.; Zhao, Z.; et al. Improving the stability of non-noble-metal M-N-C catalysts for proton-exchange-membrane fuel cells through M-N bond length and coordination regulation. Adv. Mater. 2021, 33, e2006613.
21. Wu, Y.; Ding, Y.; Han, X.; et al. Modulating coordination environment of Fe single atoms for high-efficiency all-pH-tolerated H2O2 electrochemical production. Appl. Catal. B. Environ. 2022, 315, 121578.
22. Deng, M.; Wang, D.; Li, Y. General design concept of high-performance single-atom-site catalysts for H2O2 electrosynthesis. Adv. Mater. 2024, 36, e2314340.
23. Nørskov, J. K.; Rossmeisl, J.; Logadottir, A.; et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B. 2004, 108, 17886-92.
24. Seh, Z. W.; Kibsgaard, J.; Dickens, C. F.; Chorkendorff, I.; Nørskov, J. K.; Jaramillo, T. F. Combining theory and experiment in electrocatalysis: insights into materials design. Science 2017, 355, eaad4998.
25. Chen, Y.; Ji, S.; Wang, Y.; et al. Isolated single iron atoms anchored on N-Doped porous carbon as an efficient electrocatalyst for the oxygen reduction reaction. Angew. Chem. Int. Ed. 2017, 56, 6937-41.
26. Sun, T.; Tian, B.; Lu, J.; Su, C. Recent advances in Fe (or Co)/N/C electrocatalysts for the oxygen reduction reaction in polymer electrolyte membrane fuel cells. J. Mater. Chem. A. 2017, 5, 18933-50.
27. Sun, P.; Qiao, K.; Li, D.; et al. Designing oxygen-doped Fe-N-C oxygen reduction catalysts for proton- and anion-exchange-membrane fuel cells. Chem. Catal. 2022, 2, 2750-63.
28. Gao, J.; Yang, H. B.; Huang, X.; et al. Enabling direct H2O2 production in acidic media through rational design of transition metal single atom catalyst. Chem 2020, 6, 658-74.
29. Yin, P.; Yao, T.; Wu, Y.; et al. Single cobalt atoms with precise N-coordination as superior oxygen reduction reaction catalysts. Angew. Chem. Int. Ed. Engl. 2016, 55, 10800-5.
30. Sun, Y.; Silvioli, L.; Sahraie, N. R.; et al. Activity-selectivity trends in the electrochemical production of hydrogen peroxide over single-site metal-nitrogen-carbon catalysts. J. Am. Chem. Soc. 2019, 141, 12372-81.
31. Jung, E.; Shin, H.; Lee, B. H.; et al. Atomic-level tuning of Co-N-C catalyst for high-performance electrochemical H2O2 production. Nat. Mater. 2020, 19, 436-42.
32. Tian, L.; Gao, X.; Wang, S.; et al. Precise arrangement of metal atoms at the interface by a thermal printing strategy. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2310916120.
33. Bai, X.; Wang, Y.; Han, J.; Niu, X.; Guan, J. Engineering the electronic structure of isolated manganese sites to improve the oxygen reduction, Zn-air battery and fuel cell performances. Appl. Catal. B. Environ. 2023, 337, 122966.
34. Hu, X.; Chen, S.; Chen, L.; et al. What is the Real origin of the activity of Fe-N-C electrocatalysts in the O2 reduction reaction? J. Am. Chem. Soc. 2022, 144, 18144-52.
35. Chen, S.; Luo, T.; Li, X.; et al. Identification of the highly active Co-N4 coordination motif for selective oxygen reduction to hydrogen peroxide. J. Am. Chem. Soc. 2022, 144, 14505-16.
36. Zhou, W.; Su, H.; Cheng, W.; et al. Regulating the scaling relationship for high catalytic kinetics and selectivity of the oxygen reduction reaction. Nat. Commun. 2022, 13, 6414.
37. Gao, R.; Wang, J.; Huang, Z.; et al. Pt/Fe2O3 with Pt-Fe pair sites as a catalyst for oxygen reduction with ultralow Pt loading. Nat. Energy. 2021, 6, 614-23.
38. Nishiori, D.; Menzel, J. P.; Armada, N.; et al. Breaking a molecular scaling relationship using an iron-iron fused porphyrin electrocatalyst for oxygen reduction. J. Am. Chem. Soc. 2024, 146, 11622-33.
39. Chong, L.; Wen, J.; Kubal, J.; et al. Ultralow-loading platinum-cobalt fuel cell catalysts derived from imidazolate frameworks. Science 2018, 362, 1276-81.
40. Guo, W.; Gao, X.; Zhu, M.; et al. A closely packed Pt1.5Ni1-x/Ni-N-C hybrid for relay catalysis towards oxygen reduction. Energy. Environ. Sci. 2023, 16, 148-56.
41. Wan, X.; Liu, Q.; Liu, J.; et al. Iron atom-cluster interactions increase activity and improve durability in Fe-N-C fuel cells. Nat. Commun. 2022, 13, 2963.
42. Li, L.; Yuan, K.; Chen, Y. Breaking the scaling relationship limit: from single-atom to dual-atom catalysts. Acc. Mater. Res. 2022, 3, 584-96.
43. Ying, Y.; Luo, X.; Qiao, J.; Huang, H. “More is different:” synergistic effect and structural engineering in double‐atom catalysts. Adv. Funct. Mater. 2021, 31, 2007423.
44. Yu, Z.; Xia, G.; Diaconescu, V. M.; et al. Atomically dispersed dinuclear iridium active sites for efficient and stable electrocatalytic chlorine evolution reaction. Chem. Sci. 2024, 15, 9216-23.
45. Wang, J.; Huang, Z.; Liu, W.; et al. Design of N-coordinated dual-metal sites: a stable and active Pt-free catalyst for acidic oxygen reduction reaction. J. Am. Chem. Soc. 2017, 139, 17281-4.
46. Yu, Z.; Si, C.; Lagrow, A. P.; et al. Iridium-iron diatomic active sites for efficient bifunctional oxygen electrocatalysis. ACS. Catal. 2022, 12, 9397-409.
47. Wang, X. X.; Prabhakaran, V.; He, Y.; Shao, Y.; Wu, G. Iron-free cathode catalysts for proton-exchange-membrane fuel cells: cobalt catalysts and the peroxide mitigation approach. Adv. Mater. 2019, 31, e1805126.
48. Cheng, X.; Jiang, X.; Yin, S.; et al. Instantaneous free radical scavenging by CeO2 nanoparticles adjacent to the Fe-N4 active sites for durable fuel cells. Angew. Chem. Int. Ed. Engl. 2023, 62, e202306166.
49. Xie, H.; Xie, X.; Hu, G.; et al. Ta-TiOx nanoparticles as radical scavengers to improve the durability of Fe-N-C oxygen reduction catalysts. Nat. Energy. 2022, 7, 281-9.
50. Jiang, W. J.; Gu, L.; Li, L.; et al. Understanding the high activity of Fe-N-C electrocatalysts in oxygen reduction: Fe/Fe3C nanoparticles boost the activity of Fe-Nx. J. Am. Chem. Soc. 2016, 138, 3570-8.
51. Jin, Z.; Li, P.; Meng, Y.; Fang, Z.; Xiao, D.; Yu, G. Understanding the inter-site distance effect in single-atom catalysts for oxygen electroreduction. Nat. Catal. 2021, 4, 615-22.
52. Liu, J.; Cao, C.; Liu, X.; et al. Direct observation of metal oxide nanoparticles being transformed into metal single atoms with oxygen-coordinated structure and high-loadings. Angew. Chem. Int. Ed. 2021, 60, 15248-53.
53. Hai, X.; Xi, S.; Mitchell, S.; et al. Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries. Nat. Nanotechnol. 2022, 17, 174-81.
54. Mehmood, A.; Gong, M.; Jaouen, F.; et al. High loading of single atomic iron sites in Fe-NC oxygen reduction catalysts for proton exchange membrane fuel cells. Nat. Catal. 2022, 5, 311-23.
55. Bates, J. S.; Khamespanah, F.; Cullen, D. A.; et al. Molecular catalyst synthesis strategies to prepare atomically dispersed Fe-N-C heterogeneous catalysts. J. Am. Chem. Soc. 2022, 144, 18797-802.
56. Zhang, M.; Li, H.; Chen, J.; et al. High-loading Co single atoms and clusters active sites toward enhanced electrocatalysis of oxygen reduction reaction for high-performance Zn-air battery. Adv. Funct. Mater. 2023, 33, 2209726.
57. Yin, S.; Li, Y.; Yang, J.; et al. Unveiling low temperature assembly of dense Fe-N4 active sites via hydrogenation in advanced oxygen reduction catalysts. Angew. Chem. Int. Ed. 2024, 63, e202404766.
58. Wang, Y.; Li, C.; Han, X.; et al. General negative pressure annealing approach for creating ultra-high-loading single atom catalyst libraries. Nat. Commun. 2024, 15, 5675.
59. Guo, D.; Shibuya, R.; Akiba, C.; Saji, S.; Kondo, T.; Nakamura, J. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 2016, 351, 361-5.
60. Choi, C. H.; Lim, H.; Chung, M. W.; et al. The Achilles’ heel of iron-based catalysts during oxygen reduction in an acidic medium. Energy. Environ. Sci. 2018, 11, 3176-82.
61. Liu, S.; Meyer, Q.; Jia, C.; et al. Operando deconvolution of the degradation mechanisms of iron-nitrogen-carbon catalysts in proton exchange membrane fuel cells. Energy. Environ. Sci. 2023, 16, 3792-802.
62. Osmieri, L.; Wang, G.; Cetinbas, F. C.; et al. Utilizing ink composition to tune bulk-electrode gas transport, performance, and operational robustness for a Fe-N-C catalyst in polymer electrolyte fuel cell. Nano. Energy. 2020, 75, 104943.
63. Sun, R.; Xia, Z.; Xu, X.; Deng, R.; Wang, S.; Sun, G. Periodic evolution of the ionomer/catalyst interfacial structures towards proton conductance and oxygen transport in polymer electrolyte membrane fuel cells. Nano. Energy. 2020, 75, 104919.
64. Malko, D.; Kucernak, A.; Lopes, T. In situ electrochemical quantification of active sites in Fe-N/C non-precious metal catalysts. Nat. Commun. 2016, 7, 13285.
65. Fu, X.; Zamani, P.; Choi, J. Y.; et al. In situ polymer graphenization ingrained with nanoporosity in a nitrogenous electrocatalyst boosting the performance of polymer-electrolyte-membrane fuel cells. Adv. Mater. 2017, 29.
66. Tabe, Y.; Aoyama, Y.; Kadowaki, K.; Suzuki, K.; Chikahisa, T. Impact of micro-porous layer on liquid water distribution at the catalyst layer interface and cell performance in a polymer electrolyte membrane fuel cell. J. Power. Sources. 2015, 287, 422-30.
67. Wang, S.; Chu, Y.; Lan, C.; et al. Metal-nitrogen-carbon catalysts towards acidic orr in pemfc: fundamentals, durability challenges, and improvement strategies. Chem. Synth. 2023, 3, 15.
68. Zhou, H.; Yang, T.; Kou, Z.; et al. Negative pressure pyrolysis induced highly accessible single sites dispersed on 3d graphene frameworks for enhanced oxygen reduction. Angew. Chem. Int. Ed. 2020, 59, 20465-9.
69. Liu, J.; Gong, Z.; Allen, C.; et al. Edge-hosted Fe-N3 sites on a multiscale porous carbon framework combining high intrinsic activity with efficient mass transport for oxygen reduction. Chem. Catal. 2021, 1, 1291-307.
Cite This Article
How to Cite
Guo, W.; Liu, Y.; Zhou, H.; Wu, Y. Multiscale designing principle of M-N-C towards high performance PEMFC. Microstructures 2025, 5, 2025031. http://dx.doi.org/10.20517/microstructures.2024.91
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].