


Mechanisms of cellular senescence-induced vascular aging: evidence of senotherapeutic strategies
 0
0  ,
, Abstract
Cardiovascular diseases (CVD) remain the leading cause of death worldwide, with advancing age being the primary, nonmodifiable risk factor. Vascular dysfunction, namely arterial stiffening and endothelial dysfunction, is the key antecedent to the development of clinical CVD with aging. Fundamental aging macro-mechanistic processes that drive vascular aging include excess oxidative stress, chronic inflammation, and declines in the vasodilatory molecule nitric oxide. An important hallmark of aging that contributes to the vascular aging processes is cellular senescence - a stress response characterized by cell cycle arrest and accompanied by the production and secretion of proinflammatory molecules (i.e., the senescence-associated secretory phenotype [SASP]). Excess senescent cells and the SASP have deleterious effects on vascular function and in states of CVD, making it a putative therapeutic target for improving vascular function and preventing or reversing CVD. This review will focus on the role of cellular senescence in age-related vascular dysfunction and CVD. We will examine established and emerging mechanisms underlying cellular senescence-induced vascular dysfunction. We will then discuss groups with impaired vascular function and high cellular senescence burden and examine strategies to reduce or remove excess senescent cells and the SASP in the groups who are likely to benefit most from these therapies. Finally, we will highlight the systemic effects of vascular senescent cell suppression on other tissues and organs, given the integrative role of the vasculature in physiology. Together, this review will underscore the imperative role of cellular senescence in vascular dysfunction and the need for a deeper understanding of the translational use of cellular senescence and SASP targeting therapies in groups with high senescent cell burden.

Keywords
INTRODUCTION
Cardiovascular diseases (CVD) are the leading cause of death worldwide, and despite major advances in CV medicine, advancing age remains the major CVD risk factor[1-3]. Demographic projections predict continuous increases in the number and percentage of older adults (i.e., people over 65 years of age) over the next several decades[4]. The combination of age as a risk factor for CVD and the projected increase in the number of older adults will lead to a higher incidence of CV events and a greater prevalence of CVD. As such, it is crucial to identify novel mechanisms of CVD and therapeutics to prevent and treat CVD.
Advanced age results in vascular dysfunction characterized by the stiffening of the large elastic arteries (i.e., aorta) and impaired vascular endothelial function, both of which are key antecedents of CVD[5]. Age-related vascular dysfunction is a consequence of several fundamental macro-mechanistic processes, including chronic inflammation, excess oxidative stress, and declines in the bioavailability of the vasodilatory molecule nitric oxide (NO)[6-8]. While the etiology of these fundamental macro-mechanistic processes has not been fully elucidated, the processes have been well characterized, and modulating these pathways improves vascular function in older mammals[9,10]. Cellular senescence, a hallmark of aging, has emerged as a plausible contributor to inflammation, oxidative stress, and suppressed NO[8]. Cellular senescence is a stress response characterized by stable cell cycle arrest and the production of a proinflammatory milieu referred to as the senescence-associated secretory phenotype (SASP)[11]. Advancing age increases the abundance of senescent cells in the vasculature[12], which contributes to age-related vascular dysfunction[13] and several CVD[14]. Therefore, targeting cellular senescence and the SASP using senotherapeutics - therapies that suppress excess senescent cells and/or the SASP - is an appealing strategy to treat and prevent vascular dysfunction and mitigate CVD risk[8].
In this review, we will discuss: (1) a summary of the role of cellular senescence and the SASP in vascular dysfunction; (2) established and putative mechanisms through which excess senescent cells and the SASP drive vascular dysfunction; (3) clinical groups with high senescence burden that may benefit most from senotherapeutics; and (4) translational challenges in senotherapeutic dosing and clinical implementation.
AGE-RELATED VASCULAR DYSFUNCTION AND MECHANISMS OF VASCULAR AGING
Large elastic artery stiffening and vascular endothelial dysfunction are two independent predictors of future CV events and mortality[15,16]. Stiffening of the large elastic arteries (i.e., the aorta) occurs mostly through changes in the mechanical properties of the vessel wall[17]. During aging, vascular smooth muscle cells (VSMCs) undergo phenotypic switching from synthetic to contractile states, promoting unfavorable vascular remodeling such as changes in the stress/strain components of the arteries[18]. Aortic stiffness is assessed non-invasively in mice and humans by the reference standard in vivo measure of pulse wave velocity (PWV)[19]. This assessment quantifies the stiffness of the aorta by measuring the time elapsed for the pressure wave produced by the contraction of the heart during systole to propagate bidirectionally to the two pulse pressure sites[19]. Importantly, PWV increases with advancing age and increases pressure pulsatility, thereby affecting systolic blood pressure in a feed-forward cycle[17]. As such, increased arterial stiffness contributes to hypertension and is independently predictive of future CVD[16].
Endothelial dysfunction is another primary manifestation of vascular aging characterized, in part, by impaired endothelium-dependent dilation (EDD)[20]. Endothelial cells make up a monolayer that lines the inner-most wall of blood vessels[21]. In response to mechanical shear stress induced by antegrade blood flow or exposure to pharmacological agents (i.e., acetylcholine), endothelial cells produce NO, which diffuses into the VSMC to cause vasodilation and regulate vascular tone[22]. In humans, EDD is commonly measured using the non-invasive reference-standard approach of brachial artery flow-mediated dilation (FMD), which is primarily dependent on the endothelial response to produce NO, and is a widely used measure to characterize the health of the macrovascular endothelium[15]. Another key characteristic of age-related endothelial dysfunction includes dysregulation of delivery of essential nutrients and oxygen to peripheral tissues[21]. Given that the endothelium lines the circulation, endothelial cells serve as an important barrier and regulator of biological factors that cross into peripheral tissues[21].
Fundamental macro-mechanistic processes underlying arterial stiffening and endothelial dysfunction include excess oxidative stress, chronic, low-grade inflammation, and reduced NO bioavailability[23]. With aging, oxidative stress results from a combination of excess reactive oxygen species (ROS) production and decreased antioxidant defenses due primarily to reductions in antioxidant enzyme activity/abundance[9,24]. Excess ROS readily reacts with NO to form peroxynitrite and lower NO bioavailability[24]. Peroxynitrite may also oxidize tetrahydrobiopterin (BH4), an essential cofactor for NO production by endothelial NO synthase (eNOS)[25]. Importantly, this results in eNOS uncoupling, whereby eNOS generates ROS instead of NO[25]. Together, these processes promote ROS bioactivity and reduce NO bioavailability to impair endothelial function[20]. Excess ROS may also activate proinflammatory and prooxidant signaling pathways[26]. This intrinsic inflammatory activation in the microenvironment or in the circulation can activate other ROS-producing systems, creating a feed-forward cycle of inflammation and oxidative stress that exacerbates vascular dysfunction. States of high oxidative stress and inflammation contribute to arterial stiffening by promoting adverse structural remodeling of the vascular walls[8]. Notably, four unfavorable structural changes occur in the vascular wall with aging: (1) there is a degradation and fragmentation of elastin; (2) there is a compensatory deposition of collagen to replace the fractured elastin; (3) the stimulation of advanced glycation end products (AGEs) promotes crosslinking of these structural components; and (4) AGE-induced calcium (Ca2+) deposition leading to arterial calcification[8,27]. There are also functional contributions to arterial stiffening driven by reduced vasodilatory factors (i.e., NO and hyperpolarizing factors) and increased vasoconstrictive factors (i.e., angiotensin II) that confer VSMC stiffness[8,28]. Together, these processes reduce elasticity and contribute to the stiffness of the arteries. Although these major macro-mechanistic events are well-accepted as the underlying mechanisms of vascular aging[23], the integrative cellular/molecular processes regulating excessive oxidative stress, chronic inflammation, and limited NO bioavailability with aging are incompletely understood.
THE ROLE OF CELLULAR SENESCENCE IN VASCULAR DYSFUNCTION
Cellular senescence is a complex biological process characterized by the irreversible growth arrest of cells in response to various stimuli, including DNA damage, telomere shortening, oxidative stress, and oncogene activation[11,29]. Senescent cells exhibit a distinct phenotype compared to healthy, proliferating cells, including an enlarged and flattened morphology, increased metabolic activity, and an amplified secretion of a wide range of proinflammatory cytokines, chemokines, and growth factors collectively referred to as the senescence-associated secretory phenotype (SASP)[30,31]. Senescent cells and the SASP contribute to many physiological processes, including embryonic development, wound healing, and tumor suppression[29,32]. However, the accumulation of senescent cells and the SASP has been implicated in various age-related pathologies, including CVD[33-35]. Increased cellular senescence is evident in clinical vascular pathologies including atherosclerosis[36,37], pulmonary hypertension[38], and aortic aneurysms[39]. In the absence of disease, the vasculature remains the tissue with the greatest senescent cell burden in old mice and in models of premature aging in mice[12], suggesting that the vasculature is particularly vulnerable to cellular senescence. Key vascular cells, such as endothelial cells[40-42] and VSMCs[43,44], become senescent with chronological aging and with other stressors in vivo. Importantly, vascular senescent cells are identified by key characteristics and biomarkers including cell cycle regulators (i.e., cdkn1a - p21, Cdkn2a - p16 and p19), altered lysosomal activity (senescence-associated β-galactosidase [SA-β-Gal]), DNA damage markers (53BP1 and γ-H2AX), telomere length and telomerase activity, senescent cell anti-apoptotic pathways (SCAPs), and SASP profile (IL-6, IL-1β, and tumor necrosis factor [TNF-], among many others)[31,45] [Figure 1]. Together, this evidence suggests that the burden of cellular senescence is greatest in the vasculature compared to other tissues and contributes to impairments in function and the development of CVD. Therefore, targeting senescence in the vasculature may improve function and reduce age-related vascular pathologies. Below, we discuss mechanisms of vascular cell senescence, groups with high senescent cell burden, and therapeutic treatments that target senescent cells to promote healthy vascular aging.

Figure 1. Characteristics of a vascular senescent cell. Vascular cells including endothelial cells and vascular smooth muscle cells undergo phenotypic changes upon cellular senescence induction. These senescent cell characteristics are leveraged to identify senescent cells in the vasculature.
ESTABLISHED AND EMERGING MECHANISMS UNDERLYING CELLULAR SENESCENCE-INDUCED VASCULAR DYSFUNCTION
Established mechanisms of vascular senescence
Cellular senescence has recently been causally established as a contributor to age-related vascular dysfunction. Many of the underlying mechanisms of vascular dysfunction and vascular cell senescence overlap, providing unique therapeutic targets that improve vascular function by reducing senescent cell burden. Select underlying processes have been established supporting the mechanisms by which senescent cells contribute to vascular dysfunction and serve as therapeutic targets for senescent cells[46]. Here, we discuss studies that investigate the established mechanisms by which senescent cells contribute to vascular dysfunction, and then discuss putative mechanisms that have recently emerged as novel means by which senescent cells elicit their detrimental effects on vascular physiology [Figure 2].
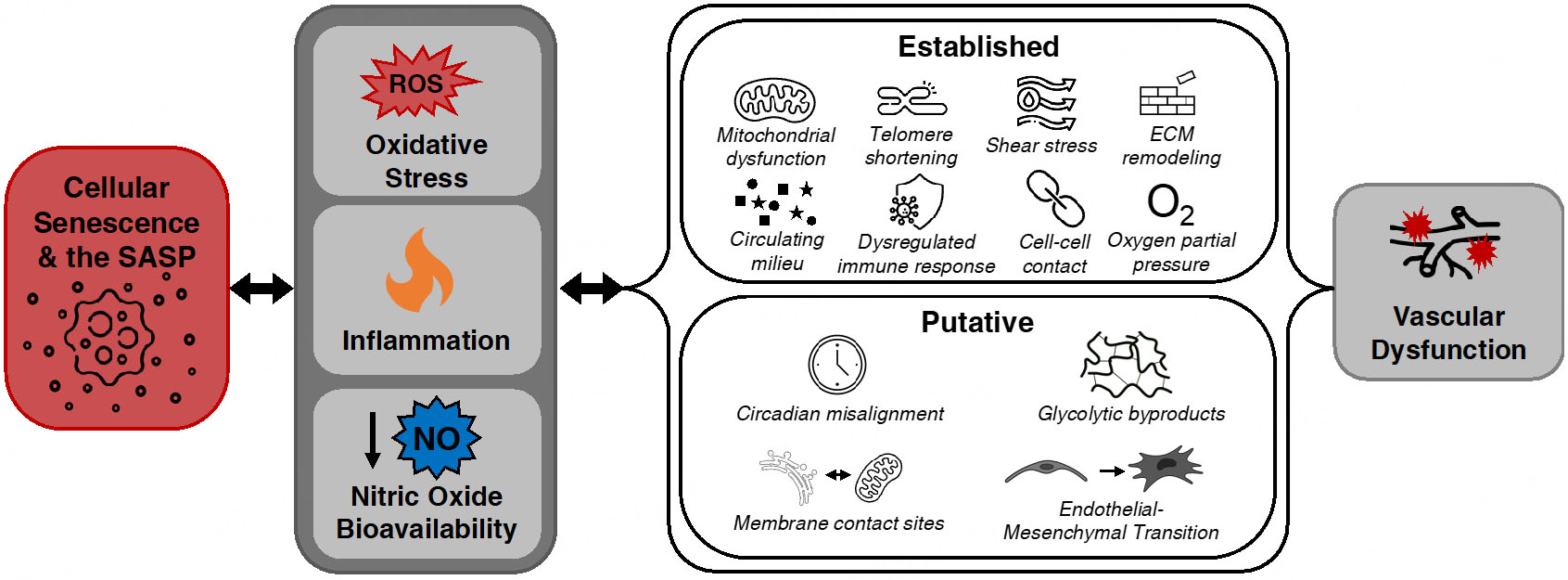
Figure 2. Mechanisms by which cellular senescence contributes to vascular dysfunction. Excess senescent cell and senescence-associated secretory phenotype (SASP) burden drive macro-mechanistic processes including reactive oxygen species (ROS)-related oxidative stress, chronic inflammation, and reduced nitric oxide (NO) bioavailability. In turn, these fundamental aging processes promote other established and putative underlying mechanisms that contribute to vascular dysfunction. ECM: Extracellular matrix; ER: endoplasmic reticulum.
Mitochondrial dysfunction
Mitochondrial dysfunction, characterized in part by reduced electron transport system function, impaired mitophagic capacity, and elevated ROS production, is a key feature of vascular aging[47]. Cellular senescence promotes mitochondrial dysfunction in both an autocrine and paracrine manner as a result of the proinflammatory SASP[48,49]. In senescent cells, mitochondria undergo outer membrane permeabilization, which releases mitochondrial DNA into the cytosol[50]. In turn, cytosolic mitochondrial DNA activates the cGAS-STING pathway, a major regulator of the SASP[50]. Mitochondrial-derived ROS contribute to vascular dysfunction with aging, as evidenced by a reduction in mitochondrial ROS, reduced vascular mitochondrial prooxidant markers, and a higher abundance of mitochondrial antioxidant defense following systemic senescent cell clearance[13,51]. Although cellular senescence and mitochondrial dysfunction are both considered hallmarks of aging, the bidirectional relationship between these processes further highlights the multiplicity of benefits of targeting senescent cells. Given the essential role of the mitochondria in senescent cells and in regulating the SASP, dysfunctional mitochondria may serve as an important therapeutic target to mitigate the detrimental effects of cellular senescence and the SASP on vascular function.
Telomere shortening and dysfunction
Telomeres, the DNA sequences and proteins that protect the caps of chromosomes, shorten with every replication cycle and are maintained by the enzyme telomerase[52]. Loss of telomere due to excess shortening or insufficient telomerase expression is an established cause of stress-induced senescence[52]. Endothelial cells have a low replicative capacity, accept at vascular regions where they are exposed to low and oscillatory shear stress, rendering them susceptible to telomere-shortening induced senescence[52]. In addition to shortening, telomere dysfunction may occur independent of length, likely due to oxidative stress to directly mediate vascular cell senescence[40]. Specifically, endothelial cell telomere dysfunction-induced senescence impairs endothelial function through increased inflammatory signaling and oxidative stress in young mice[40]. Further, telomere length is inversely associated with vascular elasticity and CVD events starting in people in mid-life[53]. More studies are warranted to determine the direct implications of telomere function/length with arterial stiffening and to extend these findings to people.
Shear stress
Endothelial cells are constantly exposed to shear stress created by blood flow. This unique environmental stimulus has pathophysiological implications, which are regional, as blood flow patterns vary widely across the vascular network. At vascular curvatures, branches, and bifurcations, low and oscillatory shear stress impairs endothelial function and is associated with an inflammatory phenotype. Conversely, high and laminar shear stress tends to be vasoprotective[54]. In relation to cellular senescence, regions of the circulation exposed to low and oscillatory shear stress exhibit elevated markers of cellular senescence and stimuli such as DNA damage and telomere dysfunction compared to cells that experience high and laminar shear stresses[40,55-57]. Importantly, these regions are also prone to the development of atherosclerosis[55,57-59]. Mechanistically, low and oscillatory shear stress induces cellular senescence by increasing cell turnover, which is likely exaggerated in the vasculature compared to other cells in the body[59,60]. However, in regions exposed to low and oscillatory shear stress, endothelial cells are pushed to divide more rapidly, and clonal hyperplasia of VSMCs contributes to atherosclerotic plaque development[61-63]. In endothelial cells of large arteries, the replicative burden is not mitigated by circulating progenitors as cell regeneration requires mitotic cell division by neighboring cells[64]. Thus, the vasculature is comprised of long-lived cells that endure long-term exposure to low, oscillatory blood flow and senescence-inducing circulating factors. At vascular bifurcations, however, replicative stress may promote states of cellular senescence.
ECM remodeling
As previously mentioned, arterial stiffening is a key pathophysiological process of vascular aging. Senescent vascular cells contribute to arterial stiffness in various capacities, including VSMC stiffness and unfavorable ECM remodeling. Particularly, senescent VSMCs have a higher secretion of ECM proteins and are stiffer and less organized than the ECM derived from young control VSMCs[27]. Pro-stiffening changes in the ECM of VSMC are mediated by altered collagen deposition and reduced contractility in senescent VSMCs[27]. Sry-box transcription factor 9 (SOX9), a regulator of chondrocyte differentiation, is a central mediator of ECM stiffness and organization in senescent VSMCs[27]. Mitochondrial-derived ROS also contributes to vascular dysfunction with aging, as evidenced by a reduction in mitochondrial ROS, reduced vascular mitochondrial prooxidant markers, and a higher abundance of mitochondrial antioxidant defense following systemic senescent cell clearance[13,51]. Importantly, senescent cell clearance results in a reduction in collagen and AGE abundance in the vascular wall of old mice[13,65]. As such, the vascular ECM is an important contributor to cellular senescence-induced arterial stiffening and a therapeutic target to prevent or reverse unfavorable vascular remodeling.
The circulating milieu
The influence of the circulating milieu (i.e., the collection of bioactive factors in systemic circulation) on physiological function has been studied using a variety of experimental approaches including heterochronic parabiosis (surgical joining of two circulatory systems)[66], heterochronic blood exchange (administration of blood/blood factors of one organism into the circulating milieu of another organism)[67], and in vitro or
Dysregulated immune responses
An additional characteristic that predisposes vascular cells to damage is their interaction with the interstitial microenvironment and neighboring cells. Inflammatory immune cells traffic through the vascular network, and endothelial cells mediate extraversion from the circulation to the extravascular space[75]. Endothelial cells can upregulate the expression of adhesion molecules and secrete inflammatory chemokines to bind and recruit immune cells, respectively, and inflammatory signals increase endothelial permeability to allow for immune infiltration to neighboring tissues[75]. Similarly, inducing cellular senescence in VSMCs increases vascular immune cell infiltration[76]. Importantly, increased senescent vascular and immune cells manifest in atherosclerotic plaques and exacerbate atherosclerotic pathology via dysregulation of immune responses exerted by the SASP[77].
Cell-cell contact
Unfavorable cell-cell contact manifests in senescent vascular cells as increased expression of adhesion molecules and reduced expression of junction proteins[78,79]. As previously mentioned, senescent endothelial cells have increased the expression of adhesion molecules on the cell surface, which attracts neighboring cells or cells in the circulation to local senescent cells[79]. Despite this improved contact, senescent cells leverage this mechanism to promote the senescent program in their microenvironment rather than for the enhancement of favorable communication[79]. In contrast, junction proteins, which are transmembrane proteins that aim to transfer material between cells, are reduced and impair cell-cell communication. Given the key role of endothelial cells in forming barriers to regulate the transport of factors from circulation to peripheral tissues, the dysregulation of junction proteins in senescent vascular cells leads to an impairment of the transport of essential nutrients and oxygen[78]. Specifically, dysfunctional barrier function in senescent endothelial cells has been observed in the brain[80], lung[81], and eye[82] and may result in dysregulated permeability of factors into these tissues[78]. As such, senescent cells contribute to the established link between increased vascular permeability and vascular dysfunction[8].
Oxygen partial pressure
Finally, it is well established that culturing cells at higher oxygen tensions hastens the onset of cellular senescence, and the partial pressure of oxygen is 97%-44%, which is greater in the vascular circulation compared to the interstitial space of other tissues[72]. The partial pressure of oxygen varies from blood to tissues to cells, and exposure to higher partial pressures of oxygen in vivo likely promotes cellular senescence through the production of ROS[83,84] produced endogenously by vascular cells, or by circulating immune cells, neighboring tissues, or through iatrogenic means.
Potential novel mechanisms of vascular senescence
As previously mentioned, the accumulation of senescent cells mediates age-related vascular dysfunction, and clearance of excess senescent cells improves vascular function in old mice[13,85]. However, the direct mechanisms by which senescent cells promote vascular aging are not fully understood. In the following section, we describe potentially novel and understudied mechanisms that could regulate cellular senescence-mediated vascular aging [Figure 2]. Although causal evidence may not exist for all these mechanisms contributing to cellular senescence-mediated vascular aging, the processes warrant mention, given that they may provide insight into future experimental considerations.
Circadian misalignment
Circadian rhythms are approximately 24-h biological oscillations that have evolved in response to predictable environmental cues[86]. All cells, including cells of the vasculature, contain precisely regulated and conserved transcriptional-translational feedback loops, which perpetuate the 24-h oscillations and ultimately regulate fundamental circadian rhythms (also referred to as clock components)[86]. Bmal1 is the only imperative clock gene required for circadian rhythm regulation[87] and is considered the master regulator of the circadian clock[88]. Moreover, the BMAL1 protein is a key transcription factor for rhythmic control of chromatin accessibility, thus influencing system-level gene expression[89]. Circadian misalignment is associated with early vascular aging in humans[90]. The physiological oscillations in Bmal1 transcription are dampened with aging[91] and knockout of Bmal1 can directly impair vascular function in young mice[92]. Moreover, in vitro evidence suggests that dampened Bmal1 oscillations contribute to the accumulation of senescent cells[93]. Thus, circadian misalignment, particularly dampened oscillations in Bmal1 transcription, may be a novel mechanism underlying cellular senescence-induced vascular aging.
Glycolytic byproducts
As mentioned above, mitochondrial dysfunction is a key characteristic of senescent cells. Mitochondrial dysfunction is often accompanied by a compensatory reliance on glycolysis for ATP production[94], and consistent with this concept, senescent cells demonstrate a preferential reliance on glycolysis[95]. Considering mitochondrial dysfunction is a well-established biological hallmark of aging[96] and dysfunctional mitochondria are directly implicated in vascular aging[47], it is warranted to investigate the role of increased glycolytic flux as a mechanism mediating cellular senescence-induced vascular aging. Serum proteomic analyses reveal that increased glycolytic metabolism is likely responsible for circulating SASP-mediated changes in vascular aging[13]. Indeed, increased glycolytic flux is implicated in vascular aging, due in part to increased production of the glycolytic byproduct and AGE precursor, methylglyoxal (MGO)[97]. Under normal physiological conditions, MGO is detoxified via the glyoxalase-1 (GLO-1) pathway; however, GLO-1 activity is reduced with aging, resulting in a greater abundance of MGO[98]. In further support of this concept, MGO has been shown to stimulate cellular senescence in human keratinocytes[99] and in the brain[100], and increasing GLO-1 activity has been demonstrated to reduce senescent cell burden in renal cells[101]. Moreover, preservation of GLO-1 activity prevents age-related vascular endothelial dysfunction[102]. Thus, investigating the role of glycolysis and MGO/GLO-1 balance in mediating cellular senescence-induced vascular aging may be valuable.
Membrane contact sites
Changes in membrane contact sites are emerging as biological mechanisms of aging, which has been reviewed in detail elsewhere[103]. Membrane contact sites comprise physical bridges that allow for the exchange of metabolites between organelles, ultimately regulating intracellular metabolic flux. Moreover, membrane contact sites create subcellular signaling platforms, which enable local protein modifications and interactions[103]. Two organelles that have garnered attention in the context of membrane contact sites are mitochondria and endoplasmic reticulum (ER), referred to herein as mitochondria-ER contacts (MERCs). MERCs are involved in the regulation of inflammation and redox signaling, and thus have been implicated in age-related pathologies, including adverse vascular remodeling[103]. Exposure of endothelial cells in culture to oxidized low-density lipoprotein, an established inducer of cellular senescence[104], results in an upregulation of MERCs[105]. Moreover, artificial tightening of MERCs in human fibroblasts leads to the induction of cellular senescence, a heightened proinflammatory secretory profile, and increased ROS production[106]. Inversely, the deletion of MERC-related proteins in senescent cells has shown to reduce overall senescence burden and favorably modulate the SASP[107,108]. These results suggest there is a clear relation between MERCs and cellular senescence, but the extent to which MERCs mediate cellular senescence (or vice versa) is not well understood. Furthermore, it remains to be determined if MERCs regulate the SASP and how MERCs and cellular senescence and the SASP interact to mediate vascular aging.
Endothelial-to-mesenchymal transition
The endothelial-to-mesenchymal transition (EndoMT) is a process by which endothelial cells lose their characteristic markers, such as VE-cadherin, CD31, TIE2, and Von Willebrand factor (VWF), while gaining increased expression of mesenchymal markers, such as alpha 2 smooth muscle actin (α-SMA), fibroblast-specific protein 1 (FSP1), and type I/III collagen, which ultimately leads to a shift toward enhanced contractile properties and reduced NO production[109]. EndoMT is commonly provoked by states of excessive inflammation and is highly implicated in vascular aging[109]; however, sources of inflammation mediating EndoMT are not fully understood. Senescent endothelial cells are more susceptible to undergoing EndoMT and the proinflammatory SASP may drive the EndoMT in non-senescent endothelial cells[110]. An emerging proinflammatory SASP factor thought to promote the endoMT is transforming growth factor-β (TGF-β), as it is a common component of the SASP[110] and an established inducer of the endoMT[109]. Although physiological levels of TGF-β are required for homeostatic endothelial function[111], excessive production of TGF-β can promote vascular aging[112,113]. As such, the EndoMT may be a mechanism by which cellular senescence and the SASP contribute to vascular aging.
GROUPS WITH HIGH CELLULAR SENESCENCE BURDEN
Cellular senescence increases with chronological age and due to other intrinsic and extrinsic stressors. As such, senescent cells accumulate disproportionally in different individuals and groups and may contribute to vascular health disparities. Conditions of early vascular aging - states in which biological age is greater than chronological age - also often demonstrate increased senescent cell accumulation[114]. In this section, we highlight select groups that may experience early vascular aging and a greater senescence burden due to chronological aging, lifestyle choices, genotoxic drugs, genetic mutations, or disease progression[114] [Figure 3].

Figure 3. Select groups with high cellular senescence burden and premature vascular aging. Groups that may have high senescent cell burden often experience early vascular aging phenotypes. HIV: Human immunodeficiency virus; CVD: cardiovascular disease.
Estrogen-deficient postmenopausal women and men with low testosterone
Aging is characterized in part by a decline in gonadal function, which presents in late life as estrogen deficiency in women and low testosterone levels in men, both of which are risk factors for the development of CVD[115]. Vascular function declines in parallel with gonadal aging in women and emerging evidence suggests this may also be the case for older adult men with low testosterone[116,117]. However, the mechanisms by which gonadal aging mediates vascular dysfunction are incompletely understood. Physiological levels of estrogen maintain homeostatic redox balance in mitochondria, and as such, estrogen deficiency can increase tonic mitochondrial ROS production. Considering mitochondrial ROS is an established inducer of cellular senescence, it is plausible that vascular aging in estrogen-deficient women is mediated by an increase in cellular senescence. Moreover, in a mouse model with accelerated senescent cell accumulation, estrogen deficiency exacerbates vascular aging[118], demonstrating an interaction between gonadal status, cellular senescence, and vascular aging. Low testosterone levels have long been associated with increased tonic ROS production and chronic low-grade inflammation; therefore, it appears likely that testosterone also regulates cellular senescence. Estrogen- and testosterone-based hormone replacement is contraindicated for CVD prevention for a variety of reasons; thus, rather than replacing hormone levels to improve vascular function in estrogen-deficient postmenopausal women and in men with low testosterone, it may be more beneficial to target the mechanisms (potentially cellular senescence) by which the reductions in these hormones drive vascular aging.
Lifestyle behaviors
Certain detrimental lifestyle behaviors may also lead to cellular senescence accumulation and early vascular aging. In preclinical models, a high-fat diet promotes vascular remodeling and stimulates vascular cell senescence[119]. This is in agreement with clinical studies observing that exercise-trained mid-life/older adults have less endothelial senescent cell burden than sedentary mid-life/older adults[42]. Additional detrimental lifestyle behaviors that have been linked to increased vascular cell senescence include cigarette smoking[120] and poor sleep quality[121]. However, more studies are needed to establish the mechanisms and implications of lifestyle behaviors on cellular senescence and early vascular aging and determine if healthy lifestyle behaviors promote healthy vascular aging.
Mental stress
Mental stress has emerged as an important risk factor for the development of CVD[122,123] as it promotes early vascular aging phenotypes[124], as reviewed in detail elsewhere[122,123,125]. In brief, there are many shared biological processes between the effects of mental stress and the mechanisms underlying age-related vascular dysfunction. Mental stress is associated with reduced NO bioavailability, impaired endothelial function, tonic elevations in ROS, and chronic low-grade inflammation[124], all of which are key mechanisms of vascular aging and features of excess cellular senescence[11,126]. Preclinical literature suggests that experimentally provoked mental stress can promote cellular senescence in the brain[127], and patients who experience frequent episodes of mental stress have higher circulating markers of cellular senescence relative to healthy controls[128]. Post-traumatic stress disorder (PTSD) is a severe type of mental stress and a psychiatric/mental condition affecting approximately 5% of the US population[129,130]. Individuals most affected by PTSD include military combat veterans, victims of assault and abuse, and those who have experienced major disasters[131]. PTSD is associated with a reduced overall and healthy lifespan, inferring PTSD accelerates biological aging[132]. As such, the relationship between PTSD and the biological mechanisms of aging has gained significant attention over the last decade. Interestingly, a comprehensive review compared SASP markers from numerous studies measuring individual factors in people with PTSD and found multiple lines of investigation suggesting that PTSD may be associated with a proinflammatory SASP profile compared to age-matched individuals without PTSD[133]. However, it remains to be determined if PTSD and mental stress directly increase the burden of cellular senescence in the vasculature and if the accumulation of senescent vascular cells is mechanistically implicated in mental stress-induced vascular aging.
Radiation
Radiation is a well-established inducer of cellular senescence that occurs in diverse settings and is most commonly experienced by cancer patients via ionizing radiation during their cancer treatment and by space travelers through high solar and cosmic radiation[134]. During radiation exposure, high-energy rays induce DNA damage, leading to the induction of cellular senescence, which has been well documented in endothelial cells and VSMCs[134,135]. Specifically, exposure of vascular cells to ionizing radiation induces cellular senescence and activates SASP-mediated inflammation[135]. In mice, ionizing radiation exposure has been shown to mediate DNA damage-induced vascular cell senescence and inflammation, and accelerated plaque formation in atherosclerotic mice[136]. Moreover, cancer survivors who received cancer treatment with ionizing radiation had 2-fold higher arterial stiffness in comparison to cancer patients who did not receive ionizing radiation[137]. Together, these data provide evidence that radiation induces vascular cell senescence and promotes early vascular aging.
Chemotherapy treatment
Common forms of cancer, including breast cancer, leukemia, and lymphomas, are often treated with anthracycline chemotherapeutic agents[138,139]. Anthracyclines are highly effective at treating cancer but are considered to be the most cardiotoxic, as patients treated with anthracyclines experience early CV-related morbidity and often die prematurely from CVD[138,139]. A key mechanism by which anthracyclines inhibit cancer growth is through the induction of cellular senescence[140], and cancer survivors previously treated with anthracyclines experience heightened (relative to age- and sex-matched healthy controls) senescent cell burden years into survivorship[141-143]. Considering the link between vascular dysfunction and overt CVD, as well as the role of cellular senescence in mediating vascular aging, it is highly plausible that cellular senescence-induced vascular aging is responsible for the early CV-related morbidity and mortality observed in these patients. Doxorubicin, the most commonly administered anthracycline chemotherapeutic agent, is a well-established inducer of cellular senescence and early vascular aging[144]. Specifically, young mice that received systemic, genetic clearance of senescent cells following doxorubicin treatment do not develop vascular dysfunction compared to doxorubicin-treated mice that did not receive genetic senescent cell clearance[145]. Other classes of chemotherapeutic agents are known to elicit cellular senescence and require further investigation into adverse effects on the vasculature[146]. Specifically, paclitaxel, a commonly used microtubule inhibitor, has direct adverse effects on the proliferation and migration of senescent vascular cells and contributes to the disruption of blood-brain barrier integrity[147,148]. Together, these data suggest that cellular senescence is likely an underlying mechanism of cancer chemotherapy-induced early vascular aging.
Antiretroviral therapy treatment
Although antiretroviral therapies are highly effective at treating the fatal effects of the sexually transmitted human immunodeficiency virus (HIV) infection, long-term use of these therapies promotes early vascular aging and accelerated senescence accumulation[149]. Antiretroviral therapies inhibit the reverse transcription utilized by HIV viruses to infect cells[150]. Like chemotherapeutic agents, antiretroviral therapies are considered to mediate stress-induced premature senescence[151]. Studies in cultured endothelial cells showed that antiretroviral therapies elicit premature senescence associated with inflammation, oxidative stress, and altered eNOS activation[152]. Additional preclinical studies in mice demonstrated that antiretroviral therapies promote states of cellular senescence by impairing mitochondrial function, defecting nuclear wall structure, and promoting the SASP, which was accompanied by physiological dysfunction[151]. In people, associations have emerged between HIV infection, antiretroviral therapy, and cellular senescence burden[150,153]. However, the direct effects of antiretroviral therapies on the vascular function of HIV-infected patients are still yet to be determined.
Vascular disease
The presence of clinical vascular disorders and associated risk factors may be another contributor to early vascular aging by increasing cellular senescence. Increased senescent cell burden has been observed in several clinical vascular disorders including atherosclerosis[154,155], pulmonary hypertension[38,156], and aortic aneurysms[157,158]; however, it is unclear whether senescent cells are primary or secondary to the onset of these conditions. Given that senescent cell accumulation is only associated with clinical vascular diseases, preclinical models are currently being leveraged to better understand the implications of cellular senescence in these conditions. Below, we highlight studies in which senescent cells play a role in vascular pathologies.
Senescent cells have been identified in atherosclerosis, a progressive disease defined by the accumulation of lipid and fibrous elements in the large elastic arteries[35]. Senescent cells exist in atherosclerotic lesions and contribute to the destabilization of plaques that may promote acute CV events[44,159]. VSMCs derived from plaques exhibit key senescent cell characteristics, including DNA damage and telomere shortening[155]. Further, SASP factors have been shown to aggravate atherosclerosis by recruiting and activating immune responses and promoting chronic inflammation and oxidative stress[160]. Pulmonary arterial hypertension is a proliferative disorder that results mainly from increased vascular cell production and contributes to CVD events and chronic lung diseases[38]. Cellular senescence contributes to pulmonary vascular remodeling, which ultimately manifests in vascular dysfunction in the lung as signified by increased right ventricular systolic pressure[38,156]. Yet, senescent cells may play a multifaceted role in the stability of the pathology by inhibiting the worsening of the pulmonary hypertension state (discussed further in this review)[38]. As such, more studies are required to determine the cost-benefit of senescent cell burden in pulmonary arterial hypertension states. Senescent cells in aortic aneurysms, characterized by a persistent, localized dilation (ballooning) of the aorta, contribute to a loss of aortic structural wall integrity, which may lead to severe CVD events[157,161]. Proteomic analyses have identified cellular senescence as a key pathway in thoracic aortic aneurysms[158]. Senescent cells also contribute to aortic aneurysm development through the secretion of proinflammatory cytokines and ROS through the SASP[161]. Senescent cells accumulate primarily at sites of vascular hypertrophy in pulmonary hypertension models[162] and contribute to the pathology through the recruitment of inflammatory cells by the SASP and by promoting vascular cell growth and migration[38,156]. Further mechanistic studies are needed to uncover the complex interaction between cellular senescence and vascular disease.
Progeria
Hutchinson-Gilford progeria syndrome (HGPS) is an extremely rare, premature aging disorder caused by the genetic mutation of the Lmna gene resulting in a defective nuclear lamina[163]. In patients with HGPS, there is a greater senescent cell burden and vascular dysfunction that manifests at young ages, with CVD being the leading cause of death in this group[164]. Mouse models of progeria have been developed to study disease progression and have also been adapted to study premature aging[12,165]. For example, the ERCC1 knockout mouse model depletes the enzyme responsible for multiple DNA repair mechanisms that protect the nuclear genome, resulting in progeria disease progression[165]. At young ages, ERCC1 knockout progeroid mice have increased cellular senescence burden in all tissues comparable to old mice, with the aorta having the greatest burden[12]. Like HGPS patients, the ERCC1 knockout progeria model exhibits impaired vascular function even at young ages[166]. However, more studies are required to determine if these vascular compromises are directly mediated by cellular senescence burden.
Another progeroid syndrome, Werner syndrome, is defined by a mutation in the Wrn gene, which encodes the WRN RecQ helicase[167]. This helicase has a variety of functions, including DNA replication and repair, telomere maintenance, and epigenetic regulation, such as heterochromatinization[167]. Unlike HGPS, Werner syndrome patients develop normally until after puberty, after which many age-related phenotypes manifest, including hair graying, sarcopenia, skin abnormalities, cataracts, atherosclerotic disease, and some cancer, with most patients succumbing to one or more malignancies in their fifth decade of life[167]. CVD is a major cause of morbidity and mortality in these patients as they demonstrate dyslipidemia, hypertension, and atherosclerosis at a very early age[168]. One of the most profound phenotypes known is the accelerated induction of cellular senescence during replicative culture collected from Werner syndrome patients[169]. In vitro models suggest that macrophages from Werner syndrome patients co-cultured with endothelial cells and VSMCs are sufficient to induce endothelial cell dysfunction and a synthetic VSMC phenotype[170]. Future studies are required to further document the direct contribution of vascular cell senescence in vascular dysfunction and CVD progression in Werner syndrome patients and in preclinical models of the disorder.
Down syndrome
Down syndrome is a genetic disorder caused by the presence of all or part of a third copy of chromosome 21 (trisomy 21; +21). It is not only associated with intellectual disability but also with a group of clinical manifestations of premature aging. These include but are not limited to Alzheimer’s disease, cancer, hearing and vision declines, and suppressed immune system function. Like progeria conditions, CVD is prevalent in people with Down syndrome and is the leading cause of death[171]. One key insight is that people with Down syndrome have blunted endothelial function compared to age-matched controls[172]. Furthermore, initial studies suggest alterations in vascular function may contribute to some of the age-related diseases in this group. Decreased cerebral blood flow has been observed prior to β amyloid accumulation in mouse models of Down syndrome[173,174]. Moreover, individuals with Down syndrome demonstrate less carotid blood flow and vascular conductance compared with individuals without Down syndrome, suggesting that alterations in vascular control mechanisms are present in Down syndrome[175]. Despite this, little is known about how vascular cell senescence and function are altered with aging in this group. While measures of cellular senescence have not been made in vascular cell types, cellular senescence markers are high in neural progenitor cells[176], primary fibroblasts[177], and primary lymphocytes[178] from people with Down syndrome. Thus, future studies should evaluate vascular cell senescence in people with Down syndrome and further explore if vascular cell senescence is a contributor to vascular dysfunction and elevated CVD risk in people with Down syndrome.
SENOTHERAPEUTICS AND TRANSLATION TO CLINICAL RESEARCH TO IMPROVE VASCULAR FUNCTION
The select groups discussed above represent groups that experience early vascular aging likely due in part to excess cellular senescence. As such, the groups stand to benefit the greatest from the clearance of excess senescent cells. In the following section, we discuss therapeutic strategies that may be leveraged to reduce vascular cell senescence and the SASP and improve vascular function. Senotherapeutics are a broad class of therapeutic strategies aimed at targeting excess senescent cells and/or the SASP[179]. Senotherapeutics often refer to natural or synthetic pharmacological agents but have also been attributed to lifestyle interventions such as aerobic exercise and weight loss in people[42,179-181]. There are two main classes of pharmacological senotherapeutics: senolytics and senomorphics[179]. Senolytic therapies are interventions that selectively suppress excess senescent cells by targeting SCAP networks[46], whereas senomorphics are compounds that suppress SASP signaling by modulating senescent cell transcription[182]. The use of senolytics or senomorphics in pathological states is challenged and highly dependent on the context (discussed in further detail later in the review)[179]. Here, we examine evidence of senotherapeutics treatment in vascular studies
Investigations of senotherapeutic agents in vascular studies
| Senotherapeutic (class) | Dosing paradigm | Model | Condition |
Senescence/
SASP outcome |
Vascular outcome | Reference |
| Navitoclax (senolytic) |
50 mg/kg/day 1 week on - 2 weeks off - 1 week on |
C57Bl/6 mice | Old age | ↓ p16 | ↑ Endothelial function ↓ Arterial stiffness |
Clayton et al., 2023[13] |
| Navitoclax (senolytic) | 100 mg/kg/day for 6 weeks or 50 mg/kg/day for 9 weeks |
Apoe KO mice | Atherosclerosis | ↓ Cdkn2a ↓ p21 |
↓ Plaques stability | Karnewar et al., 2023[186] |
| Navitoclax (senolytic) | 50 mg/kg/day 1 week on - 3 weeks off for 3 cycles |
p16-3MR Apoe KO mice | Atherosclerosis | No change Cdkn2a or SASP | ↓ Atherosclerotic plaques | Garrido et al., 2022[187] |
| Navitoclax (senolytic) | 100 mg/kg/day for 9 days | p16-3MR Ldlr KO mice | Atherosclerosis | ↓ SA-β-Gal and Cdkn2a | ↓ Atherosclerotic plaques | Childs et al., 2016[36] |
| Navitoclax (senolytic) | 100 mg/kg/day 1 week on - 3 weeks off for 3 cycles | C57Bl/6 and INK-ATTAC Ldlr KO mice | Atherosclerosis | ↓ SA-β-Gal | ↓ Atherosclerotic plaques | Childs et al., 2021[188] |
| Navitoclax (senolytic) | 10 mg/kg/day for 8 days | Monocrotaline-treated Wistar rats | Pulmonary arterial hypertension | ↓ p16 and p21 | ↓ RVSP | van der Feen et al. 2020[156] |
| Navitoclax (senolytic) | 10-50 mg/kg/day for 3 weeks | C57Bl/6 and SM22-5-HTT+ mice and Wistar rats | Pulmonary arterial hypertension | ↓ p16 and p21 ↓ DNA damage |
↑ Vascular remodeling ↓ RVSP |
Born et al., 2022[38] |
| D + Q (senolytic) |
5 mg/kg/day (D) + 10 mg/kg/day (Q) once weekly for 2 months | C57BL/6 Apoe KO mice | Old age and atherosclerosis | ↓ DNA damage | ↑ Endothelial function ↓ Arterial stiffness ↓ Atherosclerotic plaques |
Roos et al., 2016[85] |
| D + Q (senolytic) |
5 mg/kg/day D + 50 mg/kg/day Q 3 days on - 2 weeks off for 3 cycles | C57Bl/6 angiotensin II-induced mice | Old age and abdominal aortic aneurysm | ↓ Cdkn1a and SASP | ↓ Abdominal aortic aneurysm | Parvizi et al., 2021[191] |
| Fisetin (senolytic) |
100 mg/kg/day 1 week on - 2 weeks off - 1 week on |
C57Bl/6 and p16-3MR mice | Old age | ↓ p16, Cdkn2a and SCAP | ↑ Endothelial function ↓ Arterial stiffness |
Mahoney et al., 2024[65] |
| 25-Hydroxycholersterol (senolytic) | 50 mg/kg/day for 5 days | p16-3MR mice | Old age | ↓ p16, Cdkn2a and SASP | ↓ Arterial stiffness | Mahoney et al., 2023[193] |
| FOXO4-DRI (senolytic) | 5 mg/kg/day 3X per week for 3 weeks | C57Bl/6 and SM22-5-HTT+ mice | Pulmonary arterial hypertension | None reported | ↓ RVSP | Born et al., 2022[38] |
| BPTES (senolytic) | 5 g/kg/day 3X per week for 1 month | C57Bl/6 Apoe KO mice | Old age and atherosclerosis | ↓ SA-β-Gal and SASP | ↓ Atherosclerotic plaques | Johmura et al., 2026[194] |
| GPNMB vaccine (senolytic) | Not reported | C57Bl/6 Apoe KO mice | Atherosclerosis | ↓ Cdkn1a, Cdkn2a, and SA-β-Gal | ↓ Atherosclerotic plaques | Suda et al., 2022[195] |
| Pitavastatin (senomorphic) | 10-100 nmol/L once | HUVEC | H2O2-induced cellular senescence | ↓ SA-β-Gal | ↑ eNOS, SIRT1, and catalase expression | Ota et al., 2010[196] |
| Pitavastatin (senomorphic) | 3 mg/kg/day daily for entire life | Streptozotocin-treated C57Bl/6 mice | Diabetes | ↓ SA-β-Gal | ↑ eNOS, SIRT1, and catalase expression | Ota et al., 2010[196] |
| Metformin (senomorphic) | 0.5-5 mM for 24 h | HUVEC | Doxorubicin-induced senescence | ↓ SASP secretion | ↓ Endothelial adhesion molecules | Abdelgawad et al. 2023[197] |
| Exendin-4 (senomorphic) | 10 nmol/l for 72 h | HUVEC | H2O2-induced senescence | ↓ SA-β-Gal activity | None reported | Oeseburg et al. 2010[198] |
| Exendin-4 (senomorphic) | 0.01-100 nM for 72 h | Rat aortic VSMC | Angiotensin II-induced senescence | ↓ SA-β-Gal activity | ↓ ROS levels | Xhao et at. 2014[199] |
| Aspirin (senomorphic) | 100 M every 48 h until the 12th passage | HUVEC | Replicative senescence | ↓ SA-β-Gal activity ↑ Telomerase activity |
↓ ROS levels ↑ NO levels |
Bode-Böger et al., 2005[200] |
| Resveratrol (senomorphic) | 10-50 M for 72 h | HEPC | Replicative senescence | ↓ SA-β-Gal activity ↑ Telomerase activity |
↑ Cell proliferation and migration | Xia et al., 2008[201] |
| Resveratrol (senomorphic) | 1 mol/L for 48 h | Rhesus monkey VSMC | Old age | ↓ Cytokine profile ↓ SASP profile |
↓ Mitochondrial ROS production | Csiszar et al., 2011[202] |
| Metformin (senomorphic) | 0.5-5 mM for 24h | HUVEC | Doxorubicin-induced senescence | ↓ SASP secretion | ↓ Endothelial adhesion molecules | Abdelgawad et al. 2023[197] |
| Rapamycin (senomorphic) | 14 mg kg-1 for 6 weeks | B6D2F1 mice | Old age | ↓ p19 | ↑ Endothelial function and ↓ Arterial stiffness |
Lesniewski et al., 2017[203] |
| Rapamycin (senomorphic) | 10 nM for 6, 24, 48, or 72h | HCAEC | H2O2-induced senescence | ↓ SASP secretion ↓ SA-β-Gal |
↑ EndoMT transition | Sasaki et al., 2020[204] |
Senolytics
Select well-established and experimental senolytic therapies exist that suppress senescent cells by targeting SCAP networks to allow apoptosis to resume in senescent cells such that they are cleared from the tissue[183]. Senolytics therapies are often administered using a “hit-and-run” approach, which is also often referred to as an intermittent dosing paradigm[183]. This approach allows high doses of senolytics to be administered such that excess senescent cell burden can be suppressed back to basal levels. Once senescent cells reaccumulate (~2-4 weeks later in mid-life/older adults), another dosing round is administered. This approach is thought to allow for senescent cells to maintain their physiological roles (i.e., cancer suppression) while keeping levels and pathological effects to a minimum[11,29,32]. Using this dosing approach, senolytic therapies have been shown to reduce age-related senescent cell burden and improve vascular function with chronological aging, early aging, and CVD [Table 1].
Navitoclax
Initial studies investigating the role of cellular senescence in vascular dysfunction used one of the first senolytics to be established, navitoclax (also known as ABT-267 and ABT-737). Navitoclax is one of the most potent and broad-spectrum senolytic agents to be identified to date[184]. However, due to its chemotherapeutic properties, its translation to healthy mid-life/older adults is limited[185]. As such, navitoclax is primarily utilized as a “theoretical” senolytic in basic biological and preclinical studies. Preclinical studies have shown that navitoclax-induced senescent cell clearance improves endothelial function and lowers arterial stiffness in old mice[13], but may have detrimental effects on mouse models of vascular pathologies including atherosclerosis[36,186-188] and pulmonary hypertension[38,156].
Dasatinib + Quercetin
The combination senolytic treatment, D + Q, is a well-established preclinical intervention that is currently being tested in clinical populations[189,190]. Although not related to the vasculature, outcomes from the first completed clinical trials using D + Q have shown that it may reduce cellular senescence markers and select circulating SASP factors in patients with idiopathic pulmonary fibrosis[189] and Alzheimer’s disease[190]. In the vasculature, D + Q improved vasomotor function and reduced arterial stiffness in old mice[85]. Further,
Fisetin
Fisetin is a natural flavonoid with senolytic properties that is currently considered to be the safest compound for translation to mid-life/older adults[192]. In old mice, intermittent fisetin supplementation improved endothelial function, lowered arterial stiffness, and ameliorated vascular cell senescence burden[65]. Several clinical trials are underway testing the effects of fisetin supplementation on chronic conditions, including two studies that are focusing on vascular-related conditions including fisetin supplementation in healthy mid-life/older adults on vascular function (NCT06133634) and in people with peripheral artery disease (NCT06399809).
Experimental senolytics
Finally, experimental senolytic interventions, including 25-hydroxycholesterol[193], FOXO4-DRI (a peptide agonist p53 and peroxisomal membrane protein 4 [PXO4] interaction)[38], bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES; a glutaminase 1 inhibitor)[194], and a vaccine against glycoprotein nonmetastatic melanoma protein B (GPNMB; a transmembrane protein inhibitor)[195], have demonstrated initial efficacy at reducing senescent cell burden and modulating vascular function or pathological vascular disorders in preclinical models. Preliminary studies of 25-hydroxycholesterol, an endogenous cholesterol metabolite, found that vascular SCAPs were modulated to favorably modulate vascular wall remodeling and reduce arterial stiffening in old mice[193]. Notably, FOXO4-DRI worsened CV outcomes in animal models of pulmonary arterial hypertension by promoting unfavorable vascular wall remodeling[38], whereas BPTES and GPNMB interventions both had favorable effects on plaque pathology in atherosclerotic animals[194,195]. More testing is required to determine the mechanisms, safety, tolerance, and efficacy of both recognized and experimental senolytic agents in people.
Senomorphics
Established senomorphic therapies include statin, antidiabetic, and anti-inflammatory compounds, which all represent natural or FDA-approved drugs[179]. Senomorphics target SASP-mediated pathways in senescent cells by modulating central inflammatory regulators, including the transcription factor NF-κB and AKT pathway[182]. Though senomorphics are less established than senolytics, there may be contexts in which they are safer and more efficacious depending on pathological conditions (discussed in detail further in the review). Given the limited investigations of these compounds for their senomorphic properties, we will highlight the existing preclinical and cell culture evidence in vascular tissue or cells [Table 1].
Statins
Statins have various protective effects on the vasculature, including lowering cholesterol levels. However, certain protective effects may also be attributed to the inhibition of endothelial SASP production[196]. For example, pitavastatin inhibits endothelial cell senescence through AKT-dependent mechanisms in both hydrogen peroxide-induced senescence in cultured endothelial cells and streptozotocin-diabetic mice[196].
Antidiabetic drugs
Several antidiabetic drugs have been found to have beneficial effects on vascular cell function in addition to their traditional uses on glucose regulation. Of note, it is believed that metformin and glucagon-like peptide 1 (GLP-1) agonists exert some physiological benefits with aging by suppressing SASP signaling. Metformin is a potent suppressor of chronic inflammation that blocks NF-κB activity in doxorubicin-induced senescent endothelial cells[197]. Likewise, agonists of the antidiabetic hormone GLP-1, including exendin-4, may mitigate cellular senescence induction and SASP signaling in senescent endothelial cells[198] and VSMCs[199] in culture. In both cell types, exendin-4 inhibits signal transduction system that uses cyclic adenosine monophosphate (cAMP) to activate protein kinase A (PKA)[198,199]. As such, antidiabetic drugs may promote their beneficial properties in part through senomorphic effects and may be promising candidates for repurposing to more general aging populations.
Anti-inflammatory drugs
Several natural and synthetic anti-inflammatory compounds have capabilities of suppressing circulating SASP factors and modulating SASP signaling in vascular cells. Aspirin is a widely prescribed nonsteroidal anti-inflammatory drug that has also been proposed to have putative senomorphic attributions[200]. Endothelial cells in culture have delayed replicative senescence, reduced oxidative stress, and enhanced NO bioavailability following aspirin treatment[200]. Resveratrol is a natural polyphenol compound found in the skin of grapes that exerts cardioprotective effects by targeting excess oxidized low-density lipoprotein[201]. Resveratrol treatment on replicative senescent endothelial progenitor cells in culture prevents the onset of cellular senescence through activation of AKT pathways[201]. Further, VSMCs isolated from old rhesus monkeys have lower SASP production after in vitro resveratrol treatment, an effect that is mediated by an inhibition of NF-κB[202]. Finally, the immunosuppressor rapamycin and its analogs have been assessed for their senomorphic properties in senescent vascular cells. In old mice, dietary rapamycin supplementation reverses age-related vascular dysfunction while modulating cellular senescence and SASP signaling pathways[203]. Rapamycin promotes endoMT during stress-induced premature senescence through the activation of autophagy[204]. Together, assessing the senomorphic properties of these anti-inflammatory drugs enables the elucidation of the mechanisms of actions and optimal use of these compounds.
Senotherapeutic dosing
Intermittent dosing of senolytic therapies has shown success in suppressing excess senescent cells while maintaining the basal physiological function of senescent cells (i.e., cancer suppression, wound healing) in preclinical models[11,29,32]. However, limited studies have assessed the long-term effects of senolytic treatments and the number of treatment cycles has yet to be optimized. Current clinical trials testing senolytic therapies range from 2 to 6 cycles with generally 1-4 weeks of rest period in between dosing periods[189]. This dosing paradigm is also optimal for patient burden, given that medication is only administered on limited days[183]. Given the high plasticity of the vasculature (highly adaptable to change given a stimulus), senolytic therapies likely improve vascular function within a short period of time. However, tissues with less plasticity, such as the heart and skeletal muscle, may require longer dosing periods to observe improvements. Thus, future clinical trials should consider senolytic dosing paradigms based on clinical and functional outcomes [Figure 4].

Figure 4. Senotherapeutic dosing paradigms. Senolytic therapies are often administered in an intermittent dosing paradigm composed of 2-6 cycles, each involving 1-7 days of high-dose treatment, followed by a 1-4 week rest period (off treatment), allowing for the suppression of excess cellular senescence without interfering with the basal levels and functions of senescent cells (A). Senomorphic therapies are often administered continuously at a low dose to suppress excess circulating SASP and SASP signaling while maintaining basal inflammation levels (B).
Senomorphics, on the other hand, may require continuous, low-dose administration, given the constant production of the SASP. However, more preclinical studies are required to elucidate the ideal dosing paradigm[179]. Further, given the diversity of the SASP and SASP regulators, multiple senomorphic agents may need to be administered for global SASP suppression[182] [Figure 4]. Finally, more studies are required to compare senolytic versus senomorphic therapies and to determine the context in which each is more optimal.
Senotherapeutic intervention considerations in vascular disease
Although it is generally agreed upon that senescent cells contribute to several vascular diseases, senescent cell clearance is contended in select vascular pathologies and disease progression. For example, several studies have investigated the contributions and effects of the elimination of senescent cells in animal models of atherosclerosis. Although several studies found favorable effects of senolytic treatment on atherosclerotic plaques[36,85,188,194,195], two studies found that senescent cell clearance worsened outcomes, including plaque instability and rupture in advanced stages of atherosclerosis[186,187]. As such, it is hypothesized that senescent cells may contribute to atherosclerotic plaque structure and more strategies are required to understand the optimal timing for senescent cell clearance. Similar controversies exist in models of pulmonary arterial hypertension. A study by van der Feen et al. found that senescent cell clearance in a rat model of pulmonary hypertension reversed disease progression by improving vascular hemodynamics[156]. Conversely, Born et al. found that senolytics in mice and rat models of pulmonary hypertension aggravated disease severity and worsened vascular hemodynamics[38].
Finally, several senolytic compounds may have adverse safety profiles that limit their translatability to the clinic. In particular, thrombocytopenia, a condition characterized by low circulating platelet count, has been observed with Navitoclax administration[205] and D + Q administration has been reported to have several side effects including hematologic dysfunction, fluid retention and QT prolongation, and state of the extended electrical interval between the heart's contraction and relaxation[206]. These findings collectively suggest that senescent cell clearance has variable effects on vascular disease pathogenesis, and more studies are required to fully elucidate the benefits and limitations of senotherapeutic interventions in these conditions. Further, senomorphic agents may be more efficacious in contexts where senescent cell elimination is detrimental and the development of novel senotherapeutic strategies that have more specific targets may be warranted.
MULTI-ORGAN BENEFIT OF TARGETING SENESCENCE IN THE VASCULATURE
The importance of cellular senescence in the vasculature extends beyond its role in CVD. This is because cellular senescence occurs within vascular cells resident to many tissues and organs and affects neighboring non-vascular cells by disrupting blood flow regulation which is crucial for broader physiological implications [Figure 5]. Thus, studies on vascular cell senescence have been essential to our understanding of the physiological implications of cellular senescence. An increasing number of reports have identified the cells of the vasculature as some of the main cell types that becomes senescent with advancing age as well as in models of genetically and environmentally early aging. Indeed, in these settings, senescent vascular cells arise within nearly all tissues, including the eye[207,208], skeletal muscle[209], heart[210], kidney[211,212], liver[213-215], pancreas[213], testes[216], brain[217-220], adipose tissue[210,221], and lung[156,222,223]. In addition to being widespread, endothelial cells are one of the first cell types to become senescent with advancing age[214], and the cellular senescence markers p16 and p21 increase in the vasculature to a greater extent than any other tissue in both natural aging and progeroid mouse models[12]. Cumulatively, these findings support a model by which many diseases, as well as premature and healthy aging, result in vascular cell senescence that occurs early, is robustly induced, and is found within nearly every tissue. As such, an additional important benefit of targeting cellular senescence in the vasculature is that it is likely to have systemic benefits. Vascular cellular senescence directly contributes to a host of cardiometabolic diseases and cancer (reviewed in detail elsewhere[72]). Recent evidence has shed light on the mechanisms by which vascular senescence contributes to this multi-system dysfunction. This includes (1) induction of cellular senescence in neighboring cells and tissues, as well as an increase in local inflammation; and (2) dysregulation of blood flow. Through these mechanisms, inducing cellular senescence in the vasculature appears to act as a rheostat for the physiological age of an organism. This has been demonstrated by the manipulation of several known modulators of cellular senescence and the SASP in the vasculature.

Figure 5. Multi-organ benefit of targeting senescence in the vasculature. Mechanistic studies in preclinical models elucidate that the vascular senescent cells have systemic detrimental effects and clearance of senescent vascular cells elicits improvements in several tissues/organs and physiological processes.
Several examples of this phenomenon come from studies using genetically modified mice to specifically manipulate endothelial cells. For example, three independent studies have demonstrated the effects of genetic induction of telomere dysfunction specifically within endothelial cells[40,224,225]. In endothelial cells, telomere dysfunction stimulated cellular senescence and increased expression of several SASP factors[40,224,225]. Consequently, cellular senescence and the SASP are elevated in whole artery lysates, perigonadal white adipose tissue, and the liver[40,224,225]. By reducing the replicative capacity of endothelial cells, microvascular density is reduced in the gut[40] and adipose tissue vascular leak is increased[224]. Moreover, insulin-mediated production of NO is impaired[40]. In these mice, adipose tissue and the liver display elevated markers of senescence and inflammatory SASP factors and impaired glucose tolerance[40,224,225]. In addition to the inflammation in metabolically active tissues, alterations in glucose homeostasis may be a consequence of improper nutrient delivery due to a lack of vascularization and blood flow reallocation in response to metabolic stimuli[40,224,225]. Finally, running endurance and cognitive function are impaired by endothelial telomere dysfunction-induced senescence[224]. These data indicate that stimulating cellular senescence in young mice results in a premature aging phenotype that extends beyond changes typically associated with vascular dysfunction.
Mirroring the studies of telomere dysfunction in young mice, endothelial cell-specific reductions in the mammalian target of rapamycin (mTOR) reduce cellular senescence in both whole arteries and endothelial cells, as well as in metabolically active tissues including the adipose tissue and the liver[226]. Likewise, inflammatory SASP factors are reduced in these tissues[226]. These changes culminate in improvements in glucose tolerance due to a suppression of hepatic gluconeogenesis[226]. Importantly, when coupled with findings that show senescent cell clearance in old mice improves vascular function in a multitude of ways[13,65], these findings demonstrate the multiplicity of benefits of targeting cellular senescence specifically in the vasculature. It appears that senescent cells in the vasculature may promote cellular senescence and inflammation in a variety of tissues, reduce vessel density, increase vascular permeability, and disrupt proper redistribution of blood in response to metabolic stimuli, all of which act to impair metabolic function and potentially exercise tolerance and cognition. Therefore, clearing senescent vascular cells is likely to be beneficial in conditions that were not originally considered to be driven by cellular senescence in the vasculature.
RESEARCH GAPS AND FUTURE DIRECTIONS
The role of cellular senescence in vascular aging is an emerging field with promising potential in modulating clinical CV outcomes. Despite the promising evidence in the field, several research gaps exist that represent future directions in CV and cellular senescence research [Figure 6]. At a fundamental level, the mechanisms underlying vascular cell senescence and mechanisms by which senescent cells contribute to vascular dysfunction are yet to be fully elucidated. Uncovering these mechanisms may reveal novel means to target senescent cells and elucidate the detrimental effects of cellular senescence on vascular function. Next, markers of cellular senescence have only recently allowed researchers to assess cellular senescence levels in clinical populations[31,45]. As such, senescent cell burden has yet to be characterized in many clinical populations but could be a promising therapeutic target for groups that present with high levels of senescent cells. Even in the pathologies in which senescent cells have been identified, the role of the senescent cells appears to be largely heterogenous, and clearance of senescent cells has variable outcomes. Accordingly, further research exploring and optimizing the dosing, timing, and context of senotherapeutic interventions is essential for translation. Finally, several senotherapeutic strategies exist, but the safety, tolerability, and efficacy of these interventions in people are not fully known[183]. Clinical trials of the senotherapeutic compounds discussed in this review are essential, as well as the identification of novel strategies that may target specific cell types or affected areas.

Figure 6. Research gaps and future directions. Presented are important research gaps and future directions in the understanding of cellular senescence, vascular function, and cardiovascular disease (CVD). These research gaps require prioritization in ongoing and future studies.
CONCLUSIONS
Aging is the major risk factor for CVD due to the development of vascular dysfunction, particularly arterial stiffening and endothelial dysfunction. In this review, we discussed cellular senescence - a stress response characterized by cell cycle arrest - as a fundamental mechanism of vascular aging that may serve as viable therapeutic targets for the prevention and treatment of vascular dysfunction to reduce the risk of CVD. We reviewed key established and novel mechanisms by which senescent cells contribute to vascular dysfunction. We identified groups that may have high vascular cell senescence burden due to chronological or early vascular aging. Finally, we discussed senotherapeutics - therapies that suppress excess senescent cells or the SASP - and examined their translational potential, dosing paradigm, clinical implications, groups that may stand the most to benefit, and systemic benefits. Together, this review underscores the crucial role of cellular senescence in vascular dysfunction and the need for deeper understanding of the translational use of senotherapeutics in groups with high senescent cell burden.
DECLARATIONS
Acknowledgment
Figures were made using Flaticon.com (Freepik) and the licensed version of BioRender.com.
Authors’ contributions
Conceived and designed research, synthesized and interpreted data, drafted, edited, revised and approved the final manuscript: Mahoney SA, Bloom SI, Seals DR, Donato AJ, Rossman MJ, Clayton ZS
Prepared figures: Mahoney SA
Availability of data and materials
Not applicable.
Financial support and sponsorship
This study was supported by the National Institutes of Health Grants F31 HL165885 (to Mahoney SA), T32 CA009370 (to Bloom SI), K01 DK115524 (to Rossman MJ), R01 AG055822 (to Seals DR and Clayton ZS), K99 HL159241 (to Clayton ZS), R01 AG060395 (to Donato AJ), R01 AG077751 (to Donato AJ), R21/R33 AG074498 (to Donato AJ), an American Heart Association award AHA 23CDA1056582 (to Rossman MJ), VA Merit CX002211 (to Donato AJ). Support from the Nora Eccles Treadwell Foundation also supported work performed by Donato AJ.
Conflicts of interest
Donato AJ and Clayton ZS are Editorial Board members of The Journal of Cardiovascular Aging. Donato AJ and Clayton ZS were not involved in any steps of editorial processing, notably including reviewer selection, manuscript handling, and decision making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Kazi DS, Elkind MSV, Deutsch A, et al. Forecasting the economic burden of cardiovascular disease and stroke in the United States through 2050: a presidential advisory from the American heart association. Circulation. 2024;150:e89-101.
2. Martin SS, Aday AW, Almarzooq ZI, et al. 2024 heart disease and stroke statistics: a report of US and global data from the American heart association. Circulation. 2024;149:e347-913.
3. Forouzanfar MH, Alexander L, Anderson HR, et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:2287-323.
4. Olshansky SJ, Goldman DP, Zheng Y, Rowe JW. Aging in America in the twenty-first century: demographic forecasts from the MacArthur Foundation Research Network on an Aging Society. Milbank Q. 2009;87:842-62.
5. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139-46.
6. Seals DR, Kaplon RE, Gioscia-Ryan RA, LaRocca TJ. You’re only as old as your arteries: translational strategies for preserving vascular endothelial function with aging. Physiology. 2014;29:250-64.
7. Abdellatif M, Rainer PP, Sedej S, Kroemer G. Hallmarks of cardiovascular ageing. Nat Rev Cardiol. 2023;20:754-77.
8. Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of vascular aging. Circ Res. 2018;123:849-67.
9. Fleenor BS, Seals DR, Zigler ML, Sindler AL. Superoxide-lowering therapy with TEMPOL reverses arterial dysfunction with aging in mice. Aging Cell. 2012;11:269-76.
10. Lesniewski LA, Durrant JR, Connell ML, Folian BJ, Donato AJ, Seals DR. Salicylate treatment improves age-associated vascular endothelial dysfunction: potential role of nuclear factor κB and forkhead box O phosphorylation. J Gerontol A Biol Sci Med Sci. 2011;66:409-18.
11. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729-40.
12. Yousefzadeh MJ, Zhao J, Bukata C, et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell. 2020;19:e13094.
13. Clayton ZS, Rossman MJ, Mahoney SA, et al. Cellular senescence contributes to large elastic artery stiffening and endothelial dysfunction with aging: amelioration with senolytic treatment. Hypertension. 2023;80:2072-87.
14. Chen MS, Lee RT, Garbern JC. Senescence mechanisms and targets in the heart. Cardiovasc Res. 2022;118:1173-87.
15. Green DJ, Jones H, Thijssen D, Cable NT, Atkinson G. Flow-mediated dilation and cardiovascular event prediction: does nitric oxide matter?. Hypertension. 2011;57:363-9.
16. Mitchell GF, Hwang SJ, Vasan RS, et al. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010;121:505-11.
17. Chirinos JA, Segers P, Hughes T, Townsend R. Large-artery stiffness in health and disease: JACC state-of-the-art review. J Am Coll Cardiol. 2019;74:1237-63.
18. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767-801.
19. Townsend RR, Wilkinson IB, Schiffrin EL, et al. Recommendations for improving and standardizing vascular research on arterial stiffness: a scientific statement from the American Heart Association. Hypertension. 2015;66:698-722.
20. Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci. 2011;120:357-75.
21. Cahill PA, Redmond EM. Vascular endothelium - gatekeeper of vessel health. Atherosclerosis. 2016;248:97-109.
22. Harrison DG. Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest. 1997;100:2153-7.
23. Steven S, Frenis K, Oelze M, et al. Vascular inflammation and oxidative stress: major triggers for cardiovascular disease. Oxid Med Cell Longev. 2019;2019:7092151.
24. Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583-606.
25. van der Loo B, Labugger R, Skepper JN, et al. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731-44.
26. Hsieh HJ, Liu CA, Huang B, Tseng AHH, Wang DL. Shear-induced endothelial mechanotransduction: the interplay between reactive oxygen species (ROS) and nitric oxide (NO) and the pathophysiological implications. J Biomed Sci. 2014;21:3.
27. Faleeva M, Ahmad S, Theofilatos K, et al. Sox9 accelerates vascular aging by regulating extracellular matrix composition and stiffness. Circ Res. 2024;134:307-24.
28. Bellien J, Favre J, Iacob M, et al. Arterial stiffness is regulated by nitric oxide and endothelium-derived hyperpolarizing factor during changes in blood flow in humans. Hypertension. 2010;55:674-80.
30. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99-118.
31. González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021;288:56-80.
32. Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722-33.
33. Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16INK4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232-6.
34. Han Y, Kim SY. Endothelial senescence in vascular diseases: current understanding and future opportunities in senotherapeutics. Exp Mol Med. 2023;55:1-12.
35. Hu C, Zhang X, Teng T, Ma ZG, Tang QZ. Cellular senescence in cardiovascular diseases: a systematic review. Aging Dis. 2022;13:103-28.
36. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472-7.
37. Cho JH, Kim EC, Son Y, et al. CD9 induces cellular senescence and aggravates atherosclerotic plaque formation. Cell Death Differ. 2020;27:2681-96.
38. Born E, Lipskaia L, Breau M, et al. Eliminating senescent cells can promote pulmonary hypertension development and progression. Circulation. 2023;147:650-66.
39. Gao P, Gao P, Zhao J, et al. MKL1 cooperates with p38MAPK to promote vascular senescence, inflammation, and abdominal aortic aneurysm. Redox Biol. 2021;41:101903.
40. Bloom SI, Liu Y, Tucker JR, et al. Endothelial cell telomere dysfunction induces senescence and results in vascular and metabolic impairments. Aging Cell. 2023;22:e13875.
41. Jia G, Aroor AR, Jia C, Sowers JR. Endothelial cell senescence in aging-related vascular dysfunction. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1802-9.
42. Rossman MJ, Kaplon RE, Hill SD, et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. 2017;313:H890-5.
43. Chi C, Li DJ, Jiang YJ, et al. Vascular smooth muscle cell senescence and age-related diseases: state of the art. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1810-21.
44. Katsuumi G, Shimizu I, Yoshida Y, Minamino T. Vascular senescence in cardiovascular and metabolic diseases. Front Cardiovasc Med. 2018;5:18.
45. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363-7.
46. Zhu Y, Tchkonia T, Pirtskhalava T, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14:644-58.
47. Rossman MJ, Gioscia-Ryan RA, Clayton ZS, Murphy MP, Seals DR. Targeting mitochondrial fitness as a strategy for healthy vascular aging. Clin Sci. 2020;134:1491-519.
48. Miwa S, Kashyap S, Chini E, von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J Clin Invest. 2022;132:e158447.
49. Wiley CD, Velarde MC, Lecot P, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303-14.
50. Victorelli S, Salmonowicz H, Chapman J, et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature. 2023;622:627-36.
51. Mahoney SA, Dey AK, Basisty N, Herman AB. Identification and functional analysis of senescent cells in the cardiovascular system using omics approaches. Am J Physiol Heart Circ Physiol. 2023;325:H1039-58.
52. Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol (1985). 2009;106:326-32.
53. Nguyen MT, Vryer R, Ranganathan S, et al. Telomere length and vascular phenotypes in a population-based cohort of children and midlife adults. J Am Heart Assoc. 2019;8:e012707.
54. Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327-87.
55. Okuda K, Khan MY, Skurnick J, Kimura M, Aviv H, Aviv A. Telomere attrition of the human abdominal aorta: relationships with age and atherosclerosis. Atherosclerosis. 2000;152:391-8.
56. Kotla S, Vu HT, Ko KA, et al. Endothelial senescence is induced by phosphorylation and nuclear export of telomeric repeat binding factor 2-interacting protein. JCI Insight. 2019;4:124867.
57. Warboys CM, de Luca A, Amini N, et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler Thromb Vasc Biol. 2014;34:985-95.
58. VanderLaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004;24:12-22.
59. Li Y, Liu Z, Han X, et al. Dynamics of endothelial cell generation and turnover in arteries during homeostasis and diseases. Circulation. 2024;149:135-54.
60. Jones RC, Karkanias J, Krasnow MA, et al. The tabula sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376:eabl4896.
61. Jacobsen K, Lund MB, Shim J, et al. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight. 2017;2:95890.
62. Misra A, Feng Z, Chandran RR, et al. Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat Commun. 2018;9:2073.
63. Chappell J, Harman JL, Narasimhan VM, et al. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res. 2016;119:1313-23.
64. McDonald AI, Shirali AS, Aragón R, et al. Endothelial regeneration of large vessels is a biphasic process driven by local cells with distinct proliferative capacities. Cell Stem Cell. 2018;23:210-25.e6.
65. Mahoney SA, Venkatasubramanian R, Darrah MA, et al. Intermittent supplementation with fisetin improves arterial function in old mice by decreasing cellular senescence. Aging Cell. 2024;23:e14060.
66. Conboy MJ, Conboy IM, Rando TA. Heterochronic parabiosis: historical perspective and methodological considerations for studies of aging and longevity. Aging Cell. 2013;12:525-30.
67. Jeon OH, Mehdipour M, Gil TH, et al. Systemic induction of senescence in young mice after single heterochronic blood exchange. Nat Metab. 2022;4:995-1006.
68. Craighead DH, Heinbockel TC, Freeberg KA, et al. Time-efficient inspiratory muscle strength training lowers blood pressure and improves endothelial function, NO bioavailability, and oxidative stress in midlife/older adults with above-normal blood pressure. J Am Heart Assoc. 2021;10:e020980.
69. Rossman MJ, Gioscia-Ryan RA, Santos-Parker JR, et al. Inorganic nitrite supplementation improves endothelial function with aging: translational evidence for suppression of mitochondria-derived oxidative stress. Hypertension. 2021;77:1212-22.
70. Mahoney SA, VanDongen NS, Greenberg NT, et al. Role of the circulating milieu in age-related arterial dysfunction: a novel ex vivo approach. Am J Physiol Heart Circ Physiol. 2024;326:H1279-90.
71. Kiss T, Tarantini S, Csipo T, et al. Circulating anti-geronic factors from heterochonic parabionts promote vascular rejuvenation in aged mice: transcriptional footprint of mitochondrial protection, attenuation of oxidative stress, and rescue of endothelial function by young blood. Geroscience. 2020;42:727-48.
72. Bloom SI, Islam MT, Lesniewski LA, Donato AJ. Mechanisms and consequences of endothelial cell senescence. Nat Rev Cardiol. 2023;20:38-51.
73. Lee AH, Ghosh D, Koh IL, Dawson MR. Senescence-associated exosomes transfer miRNA-induced fibrosis to neighboring cells. Aging. 2023;15:1237-56.
74. Terlecki-Zaniewicz L, Lämmermann I, Latreille J, et al. Small extracellular vesicles and their miRNA cargo are anti-apoptotic members of the senescence-associated secretory phenotype. Aging. 2018;10:1103-32.
75. Amersfoort J, Eelen G, Carmeliet P. Immunomodulation by endothelial cells - partnering up with the immune system?. Nat Rev Immunol. 2022;22:576-88.
76. Uryga AK, Grootaert MOJ, Garrido AM, et al. Telomere damage promotes vascular smooth muscle cell senescence and immune cell recruitment after vessel injury. Commun Biol. 2021;4:611.
77. Mazan-Mamczarz K, Tsitsipatis D, Carr A, et al. Single-cell and spatial transcriptomics uncovers the role of senescent vascular cells in pathological arterial remodeling during atherosclerosis. Research Square Platform LLC; 2023.
78. Krouwer VJ, Hekking LH, Langelaar-Makkinje M, Regan-Klapisz E, Post JA. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc Cell. 2012;4:12.
79. Mun GI, Boo YC. Identification of CD44 as a senescence-induced cell adhesion gene responsible for the enhanced monocyte recruitment to senescent endothelial cells. Am J Physiol Heart Circ Physiol. 2010;298:H2102-11.
80. Real MGC, Falcione SR, Boghozian R, et al. Endothelial cell senescence effect on the blood-brain barrier in stroke and cognitive impairment. Neurology. 2024;103:e210063.
81. Beidokhti M, Villalba N, Ma Y, Reynolds A, Villamil JH, Yuan SY. Lung endothelial cell senescence impairs barrier function and promotes neutrophil adhesion and migration. Geroscience. 2025. Online ahead of print.
82. Tang L, Zhang C, Yang Q, et al. Melatonin maintains inner blood-retinal barrier via inhibition of p38/TXNIP/NF-κB pathway in diabetic retinopathy. J Cell Physiol. 2021;236:5848-64.
83. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741-7.
84. Busuttil RA, Rubio M, Dollé ME, Campisi J, Vijg J. Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell. 2003;2:287-94.
85. Roos CM, Zhang B, Palmer AK, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15:973-7.
86. Ko CH, Takahashi JS. Molecular components of the mammalian circadian clock. Hum Mol Genet. 2006;15:R271-7.
87. Bunger MK, Wilsbacher LD, Moran SM, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009-17.
88. Rey G, Cesbron F, Rougemont J, Reinke H, Brunner M, Naef F. Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol. 2011;9:e1000595.
89. Menet JS, Pescatore S, Rosbash M. CLOCK:BMAL1 is a pioneer-like transcription factor. Genes Dev. 2014;28:8-13.
90. Crnko S, Cour M, Van Laake LW, Lecour S. Vasculature on the clock: Circadian rhythm and vascular dysfunction. Vascul Pharmacol. 2018;108:1-7.
91. Fan R, Peng X, Xie L, et al. Importance of bmal1 in Alzheimer's disease and associated aging-related diseases: mechanisms and interventions. Aging Cell. 2022;21:e13704.
92. Bhatwadekar AD, Beli E, Diao Y, et al. Conditional deletion of bmal1 accentuates microvascular and macrovascular injury. Am J Pathol. 2017;187:1426-35.
93. Jachim SK, Zhong J, Ordog T, et al. BMAL1 modulates senescence programming via AP-1. Aging. 2023;15:9984-10009.
94. Rafikov R, Sun X, Rafikova O, et al. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biol. 2015;6:278-86.
95. Wiley CD, Campisi J. From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab. 2016;23:1013-21.
96. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186:243-78.
97. der Bruggen MM, Spronck B, Delhaas T, Reesink KD, Schalkwijk CG. The putative role of methylglyoxal in arterial stiffening: a review. Heart Lung Circ. 2021;30:1681-93.
98. Aragonès G, Rowan S, Francisco SG, et al. The glyoxalase system in age-related diseases: nutritional intervention as anti-ageing strategy. Cells. 2021;10:1852.
99. Halkoum R, Salnot V, Capallere C, et al. Glyoxal induces senescence in human keratinocytes through oxidative stress and activation of the protein kinase B/FOXO3a/p27KIP1 pathway. J Invest Dermatol. 2022;142:2068-78.e7.
100. Li H, Zheng L, Chen C, Liu X, Zhang W. Brain senescence caused by elevated levels of reactive metabolite methylglyoxal on D-galactose-induced aging mice. Front Neurosci. 2019;13:1004.
101. Ikeda Y, Inagi R, Miyata T, et al. Glyoxalase I retards renal senescence. Am J Pathol. 2011;179:2810-21.
102. Jo-Watanabe A, Ohse T, Nishimatsu H, et al. Glyoxalase I reduces glycative and oxidative stress and prevents age-related endothelial dysfunction through modulation of endothelial nitric oxide synthase phosphorylation. Aging Cell. 2014;13:519-28.
103. Ziegler DV, Martin N, Bernard D. Cellular senescence links mitochondria-ER contacts and aging. Commun Biol. 2021;4:1323.
104. Wang J, Bai Y, Zhao X, et al. oxLDL-mediated cellular senescence is associated with increased NADPH oxidase p47phox recruitment to caveolae. Biosci Rep. 2018;38:BSR20180283.
105. Yu S, Zhang L, Liu C, Yang J, Zhang J, Huang L. PACS2 is required for ox-LDL-induced endothelial cell apoptosis by regulating mitochondria-associated ER membrane formation and mitochondrial Ca2+ elevation. Exp Cell Res. 2019;379:191-202.
106. Ziegler DV, Vindrieux D, Goehrig D, et al. Calcium channel ITPR2 and mitochondria-ER contacts promote cellular senescence and aging. Nat Commun. 2021;12:720.
107. Bolinches-Amorós A, Mollá B, Pla-Martín D, Palau F, González-Cabo P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front Cell Neurosci. 2014;8:124.
108. Rodríguez LR, Calap-Quintana P, Lapeña-Luzón T, et al. Oxidative stress modulates rearrangement of endoplasmic reticulum-mitochondria contacts and calcium dysregulation in a Friedreich's ataxia model. Redox Biol. 2020;37:101762.
109. Piera-Velazquez S, Jimenez SA. Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol Rev. 2019;99:1281-324.
110. Ramadhiani R, Ikeda K, Hirata KI, Emoto N. Endothelial cell senescence exacerbates pulmonary fibrosis potentially through accelerated endothelial to mesenchymal transition. Kobe J Med Sci. 2021;67:E84-91.
111. Zhu J, Angelov S, Alp Yildirim I, et al. Loss of transforming growth factor beta signaling in aortic smooth muscle cells causes endothelial dysfunction and aortic hypercontractility. Arterioscler Thromb Vasc Biol. 2021;41:1956-71.
112. Fleenor BS, Marshall KD, Durrant JR, Lesniewski LA, Seals DR. Arterial stiffening with ageing is associated with transforming growth factor-β1-related changes in adventitial collagen: reversal by aerobic exercise. J Physiol. 2010;588:3971-82.
113. Fleenor BS, Marshall KD, Rippe C, Seals DR. Replicative aging induces endothelial to mesenchymal transition in human aortic endothelial cells: potential role of inflammation. J Vasc Res. 2012;49:59-64.
114. Hamczyk MR, Nevado RM, Barettino A, Fuster V, Andrés V. Biological versus chronological aging: JACC focus seminar. J Am Coll Cardiol. 2020;75:919-30.
115. Sharma R, Oni OA, Gupta K, et al. Normalization of testosterone level is associated with reduced incidence of myocardial infarction and mortality in men. Eur Heart J. 2015;36:2706-15.
116. Vlachopoulos C, Ioakeimidis N, Miner M, et al. Testosterone deficiency: a determinant of aortic stiffness in men. Atherosclerosis. 2014;233:278-83.
117. Yildiz O, Seyrek M. Vasodilating mechanisms of testosterone. Exp Clin Endocrinol Diabetes. 2007;115:1-6.
118. Novella S, Dantas AP, Segarra G, et al. Gathering of aging and estrogen withdrawal in vascular dysfunction of senescent accelerated mice. Exp Gerontol. 2010;45:868-74.
119. Maeda M, Hayashi T, Mizuno N, Hattori Y, Kuzuya M. Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: role of superoxide production by NADPH oxidase. PLoS One. 2015;10:e0123169.
120. Farhat N, Thorin-Trescases N, Voghel G, et al. Stress-induced senescence predominates in endothelial cells isolated from atherosclerotic chronic smokers. Can J Physiol Pharmacol. 2008;86:761-9.
121. Carreras A, Zhang SX, Peris E, et al. Chronic sleep fragmentation induces endothelial dysfunction and structural vascular changes in mice. Sleep. 2014;37:1817-24.
122. Satyjeet F, Naz S, Kumar V, et al. Psychological stress as a risk factor for cardiovascular disease: a case-control study. Cureus. 2020;12:e10757.
123. Vancheri F, Longo G, Vancheri E, Henein MY. Mental stress and cardiovascular health-part I. J Clin Med. 2022;11:3353.
124. Sara JDS, Toya T, Ahmad A, et al. Mental stress and its effects on vascular health. Mayo Clin Proc. 2022;97:951-90.
125. Sara JDS, Lerman LO, Lerman A. What can biologic aging tell us about the effects of mental stress on vascular health. Hypertension. 2023;80:2515-22.
126. Hayashi T, Yano K, Matsui-Hirai H, Yokoo H, Hattori Y, Iguchi A. Nitric oxide and endothelial cellular senescence. Pharmacol Ther. 2008;120:333-9.
127. Lin YF, Wang LY, Chen CS, Li CC, Hsiao YH. Cellular senescence as a driver of cognitive decline triggered by chronic unpredictable stress. Neurobiol Stress. 2021;15:100341.
128. Rentscher KE, Carroll JE, Repetti RL, Cole SW, Reynolds BM, Robles TF. Chronic stress exposure and daily stress appraisals relate to biological aging marker p16INK4a. Psychoneuroendocrinology. 2019;102:139-48.
129. Jimenez DE, Alegría M, Chen CN, Chan D, Laderman M. Prevalence of psychiatric illnesses in older ethnic minority adults. J Am Geriatr Soc. 2010;58:256-64.
130. Pietrzak RH, Goldstein RB, Southwick SM, Grant BF. Prevalence and axis I comorbidity of full and partial posttraumatic stress disorder in the United States: results from wave 2 of the national epidemiologic survey on alcohol and related conditions. J Anxiety Disord. 2011;25:456-65.
131. Kaiser A, Cook JM, Glick DM, Moye J. Posttraumatic stress disorder in older adults: a conceptual review. Clin Gerontol. 2019;42:359-76.
132. Wolf EJ, Morrison FG. Traumatic stress and accelerated cellular aging: from epigenetics to cardiometabolic disease. Curr Psychiatry Rep. 2017;19:75.
133. Lohr JB, Palmer BW, Eidt CA, et al. Is post-traumatic stress disorder associated with premature senescence? A review of the literature. Am J Geriatr Psychiatry. 2015;23:709-25.
134. Wang Y, Boerma M, Zhou D. Ionizing radiation-induced endothelial cell senescence and cardiovascular diseases. Radiat Res. 2016;186:153-61.
135. Zheng X, Liu Z, Bin Y, et al. Ionizing radiation induces vascular smooth muscle cell senescence through activating NF-κB/CTCF/p16 pathway. Biochim Biophys Acta Mol Basis Dis. 2024;1870:166994.
136. Yamamoto Y, Minami M, Yoshida K, et al. Irradiation accelerates plaque formation and cellular senescence in flow-altered carotid arteries of apolipoprotein E knock-out mice. J Am Heart Assoc. 2021;10:e020712.
137. Fernández-Alvarez V, Nieto CS, Alvarez FL. Arterial stiffness as an ultrasound biomarker of radiation-induced carotid artery disease. Vasa. 2021;50:348-55.
138. Bansal N, Amdani SM, Hutchins KK, Lipshultz SE. Cardiovascular disease in survivors of childhood cancer. Curr Opin Pediatr. 2018;30:628-38.
139. Lipshultz SE, Karnik R, Sambatakos P, Franco VI, Ross SW, Miller TL. Anthracycline-related cardiotoxicity in childhood cancer survivors. Curr Opin Cardiol. 2014;29:103-12.
140. Cappetta D, Rossi F, Piegari E, et al. Doxorubicin targets multiple players: a new view of an old problem. Pharmacol Res. 2018;127:4-14.
141. Bourlon MT, Velazquez HE, Hinojosa J, et al. Immunosenescence profile and expression of the aging biomarker (p16INK4a) in testicular cancer survivors treated with chemotherapy. BMC Cancer. 2020;20:882.
142. Smitherman AB, Wood WA, Mitin N, et al. Accelerated aging among childhood, adolescent, and young adult cancer survivors is evidenced by increased expression of p16INK4a and frailty. Cancer. 2020;126:4975-83.
143. Muss HB, Smitherman A, Wood WA, et al. p16 a biomarker of aging and tolerance for cancer therapy. Transl Cancer Res. 2020;9:5732-42.
144. Clayton ZS, Hutton DA, Mahoney SA, Seals DR. Anthracycline chemotherapy-mediated vascular dysfunction as a model of accelerated vascular aging. Aging Cancer. 2021;2:45-69.
145. Hutton D, Brunt V, Mahoney S, et al. Cellular senescence mediates doxorubicin-induced arterial dysfunction via activation of mitochondrial oxidative stress and the mammalian target of rapamycin. FASEB J. 2021;35:00283.
146. Jain D, Ahmad T, Cairo M, Aronow W. Cardiotoxicity of cancer chemotherapy: identification, prevention and treatment. Ann Transl Med. 2017;5:348.
147. Axel DI, Kunert W, Göggelmann C, et al. Paclitaxel inhibits arterial smooth muscle cell proliferation and migration in vitro and in vivo using local drug delivery. Circulation. 1997;96:636-45.
148. Ahire C, Nyul-Toth A, DelFavero J, et al. Accelerated cerebromicrovascular senescence contributes to cognitive decline in a mouse model of paclitaxel (Taxol)-induced chemobrain. Aging Cell. 2023;22:e13832.
149. Kanmogne GD. HIV Infection, antiretroviral drugs, and the vascular endothelium. Cells. 2024;13:672.
150. Gianesin K, Noguera-Julian A, Zanchetta M, et al. Premature aging and immune senescence in HIV-infected children. AIDS. 2016;30:1363-73.
151. Kuehnemann C, Hughes JB, Desprez PY, Melov S, Wiley CD, Campisi J. Antiretroviral protease inhibitors induce features of cellular senescence that are reversible upon drug removal. Aging Cell. 2023;22:e13750.
152. Cohen J, D'Agostino L, Tuzer F, Torres C. HIV antiretroviral therapy drugs induce premature senescence and altered physiology in HUVECs. Mech Ageing Dev. 2018;175:74-82.
153. Kaur G, Sohanur Rahman M, Shaikh S, et al. Emerging roles of senolytics/senomorphics in HIV-related co-morbidities. Biochem Pharmacol. 2024;228:116179.
154. Matthews C, Gorenne I, Scott S, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99:156-64.
155. Wang J, Uryga AK, Reinhold J, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132:1909-19.
156. van der Feen DE, Bossers GPL, Hagdorn QAJ, et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci Transl Med. 2020;12:eaaw4974.
157. Chen HZ, Wang F, Gao P, et al. Age-associated sirtuin 1 reduction in vascular smooth muscle links vascular senescence and inflammation to abdominal aortic aneurysm. Circ Res. 2016;119:1076-88.
158. Tyrrell DJ, Chen J, Li BY, et al. Aging alters the aortic proteome in health and thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol. 2022;42:1060-76.
159. Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15-26.
160. Sun Y, Wang X, Liu T, Zhu X, Pan X. The multifaceted role of the SASP in atherosclerosis: from mechanisms to therapeutic opportunities. Cell Biosci. 2022;12:74.
161. Lu H, Du W, Ren L, et al. Vascular smooth muscle cells in aortic aneurysm: from genetics to mechanisms. J Am Heart Assoc. 2021;10:e023601.
162. Noureddine H, Gary-Bobo G, Alifano M, et al. Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circ Res. 2011;109:543-53.
163. Gonzalo S, Kreienkamp R, Askjaer P. Hutchinson-gilford progeria syndrome: a premature aging disease caused by LMNA gene mutations. Ageing Res Rev. 2017;33:18-29.
164. Xu Q, Mojiri A, Boulahouache L, Morales E, Walther BK, Cooke JP. Vascular senescence in progeria: role of endothelial dysfunction. Eur Heart J Open. 2022;2:oeac047.
165. Wong A, Kieu T, Robbins PD. The Ercc1-/Δ mouse model of accelerated senescence and aging for identification and testing of novel senotherapeutic interventions. Aging. 2020;12:24481-3.
166. Ataei Ataabadi E, Golshiri K, van der Linden J, et al. Vascular ageing features caused by selective DNA damage in smooth muscle cell. Oxid Med Cell Longev. 2021;2021:2308317.
167. Shamanna RA, Croteau DL, Lee JH, Bohr VA. Recent advances in understanding werner syndrome. F1000Res. 2017;6:1779.
168. Kato H, Maezawa Y. Atherosclerosis and cardiovascular diseases in progeroid syndromes. J Atheroscler Thromb. 2022;29:439-47.
169. Norwood TH, Hoehn H, Salk D, Martin GM. Cellular aging in Werner's syndrome: a unique phenotype?. J Invest Dermatol. 1979;73:92-6.
170. Paul SK, Oshima M, Patil A, et al. Retrotransposons in Werner syndrome-derived macrophages trigger type I interferon-dependent inflammation in an atherosclerosis model. Nat Commun. 2024;15:4772.
171. Zhu JL, Hasle H, Correa A, et al. Survival among people with down syndrome: a nationwide population-based study in denmark. Genet Med. 2013;15:64-9.
172. Cappelli-Bigazzi M, Santoro G, Battaglia C, et al. Endothelial cell function in patients with down's syndrome. Am J Cardiol. 2004;94:392-5.
173. Koike MA, Green KN, Blurton-Jones M, Laferla FM. Oligemic hypoperfusion differentially affects tau and amyloid-β. Am J Pathol. 2010;177:300-10.
174. Garcia-Alloza M, Gregory J, Kuchibhotla KV, et al. Cerebrovascular lesions induce transient β-amyloid deposition. Brain. 2011;134:3697-707.
175. Wee SO, Rosenberg AJ, Kanokwan B, Griffith G, Baynard T, Fernhall B. Carotid vascular blood flow in individuals with down syndrome following low body negative pressure challenge. FASEB J. 2017;31:840.21.
176. Meharena HS, Marco A, Dileep V, et al. Down-syndrome-induced senescence disrupts the nuclear architecture of neural progenitors. Cell Stem Cell. 2022;29:116-30.e7.
177. Gimeno A, García-Giménez JL, Audí L, et al. Decreased cell proliferation and higher oxidative stress in fibroblasts from Down Syndrome fetuses. Preliminary study. Biochim Biophys Acta. 2014;1842:116-25.
178. de Arruda Cardoso Smith M, Borsatto-Galera B, Feller RI, et al. Telomeres on chromosome 21 and aging in lymphocytes and gingival fibroblasts from individuals with Down syndrome. J Oral Sci. 2004;46:171-7.
179. Zhang L, Pitcher LE, Prahalad V, Niedernhofer LJ, Robbins PD. Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. FEBS J. 2023;290:1362-83.
180. Chen XK, Yi ZN, Wong GTC, et al. Is exercise a senolytic medicine? A systematic review. Aging Cell. 2021;20:e13294.
181. Lynch DH, Petersen CL, Stewart D, et al. Changes in senescence markers after a weight loss intervention in older adults with obesity. Arch Gerontol Geriatr. 2025;129:105685.
182. Wang B, Han J, Elisseeff JH, Demaria M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat Rev Mol Cell Biol. 2024;25:958-78.
183. Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med. 2020;288:518-36.
184. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15:428-35.
185. Wilson WH, O'Connor OA, Czuczman MS, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149-59.
186. Karnewar S, Karnewar V, Shankman LS, Owens GK. Treatment of advanced atherosclerotic mice with ABT-263 reduced indices of plaque stability and increased mortality. JCI Insight. 2024;9:e173863.
187. Garrido AM, Kaistha A, Uryga AK, et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc Res. 2022;118:1713-27.
188. Childs BG, Zhang C, Shuja F, et al. Senescent cells suppress innate smooth muscle cell repair functions in atherosclerosis. Nat Aging. 2021;1:698-714.
189. Justice JN, Nambiar AM, Tchkonia T, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;40:554-63.
190. Gonzales MM, Garbarino VR, Kautz T, et al. Senolytic therapy to modulate the progression of Alzheimer's disease (SToMP-AD) - outcomes from the first clinical trial of senolytic therapy for Alzheimer's disease. Res Sq. 2023;rs.3.rs-2809973.
191. Parvizi M, Franchi F, Arendt BK, Ebtehaj S, Rodriguez-Porcel M, Lanza IR. Senolytic agents lessen the severity of abdominal aortic aneurysm in aged mice. Exp Gerontol. 2021;151:111416.
192. Yousefzadeh MJ, Zhu Y, McGowan SJ, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 2018;36:18-28.
193. Mahoney S, Ciotlos S, Darrah M, et al. 25-Hydroxycholesterol reduces aortic cellular senescence and stiffness in old mice. Physiology. 2023;38:5711505.
194. Johmura Y, Yamanaka T, Omori S, et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science. 2021;371:265-70.
195. Suda M, Shimizu I, Katsuumi G, et al. Senolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat Aging. 2021;1:1117-26.
196. Ota H, Eto M, Kano MR, et al. Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler Thromb Vasc Biol. 2010;30:2205-11.
197. Abdelgawad IY, Agostinucci K, Sadaf B, Grant MKO, Zordoky BN. Metformin mitigates SASP secretion and LPS-triggered hyper-inflammation in Doxorubicin-induced senescent endothelial cells. Front Aging. 2023;4:1170434.
198. Oeseburg H, de Boer RA, Buikema H, van der Harst P, van Gilst WH, Silljé HH. Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase A. Arterioscler Thromb Vasc Biol. 2010;30:1407-14.
199. Zhao L, Li AQ, Zhou TF, Zhang MQ, Qin XM. Exendin-4 alleviates angiotensin II-induced senescence in vascular smooth muscle cells by inhibiting Rac1 activation via a cAMP/PKA-dependent pathway. Am J Physiol Cell Physiol. 2014;307:C1130-41.
200. Bode-Böger SM, Martens-Lobenhoffer J, Täger M, Schröder H, Scalera F. Aspirin reduces endothelial cell senescence. Biochem Biophys Res Commun. 2005;334:1226-32.
201. Xia L, Wang XX, Hu XS, et al. Resveratrol reduces endothelial progenitor cells senescence through augmentation of telomerase activity by Akt-dependent mechanisms. Br J Pharmacol. 2008;155:387-94.
202. Csiszar A, Sosnowska D, Wang M, Lakatta EG, Sonntag WE, Ungvari Z. Age-associated proinflammatory secretory phenotype in vascular smooth muscle cells from the non-human primate Macaca mulatta: reversal by resveratrol treatment. J Gerontol A Biol Sci Med Sci. 2012;67:811-20.
203. Lesniewski LA, Seals DR, Walker AE, et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell. 2017;16:17-26.
204. Sasaki N, Itakura Y, Toyoda M. Rapamycin promotes endothelial-mesenchymal transition during stress-induced premature senescence through the activation of autophagy. Cell Commun Signal. 2020;18:43.
205. Schoenwaelder SM, Jarman KE, Gardiner EE, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118:1663-74.
206. Breccia M, Molica M, Alimena G. How tyrosine kinase inhibitors impair metabolism and endocrine system function: a systematic updated review. Leuk Res. 2014;38:1392-8.
207. Crespo-Garcia S, Tsuruda PR, Dejda A, et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab. 2021;33:818-32.e7.
208. Crespo-Garcia S, Fournier F, Diaz-Marin R, et al. Therapeutic targeting of cellular senescence in diabetic macular edema: preclinical and phase 1 trial results. Nat Med. 2024;30:443-54.
209. Moiseeva V, Cisneros A, Sica V, et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature. 2023;613:169-78.
210. Wang B, Wang L, Gasek NS, et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat Aging. 2021;1:962-73.
211. Li M, Wang D, Liu Z, et al. Assessing the effects of aging on the renal endothelial cell landscape using single-cell RNA sequencing. Front Genet. 2023;14:1175716.
212. Cohen C, Le Goff O, Soysouvanh F, et al. Glomerular endothelial cell senescence drives age-related kidney disease through PAI-1. EMBO Mol Med. 2021;13:e14146.
213. Amor C, Fernández-Maestre I, Chowdhury S, et al. Prophylactic and long-lasting efficacy of senolytic CAR T cells against age-related metabolic dysfunction. Nat Aging. 2024;4:336-49.
214. Grosse L, Wagner N, Emelyanov A, et al. Defined p16High senescent cell types are indispensable for mouse healthspan. Cell Metab. 2020;32:87-99.e6.
215. Omori S, Wang TW, Johmura Y, et al. Generation of a p16 reporter mouse and its use to characterize and target p16High cells in vivo. Cell Metab. 2020;32:814-28.e6.
216. Ozawa M, Mori H, Endo T, et al. Age-related decline in spermatogenic activity accompanied with endothelial cell senescence in male mice. iScience. 2023;26:108456.
217. Kiss T, Nyúl-Tóth Á, Balasubramanian P, et al. Single-cell RNA sequencing identifies senescent cerebromicrovascular endothelial cells in the aged mouse brain. Geroscience. 2020;42:429-44.
218. Ximerakis M, Lipnick SL, Innes BT, et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci. 2019;22:1696-708.
219. Hussong SA, Banh AQ, Van Skike CE, et al. Soluble pathogenic tau enters brain vascular endothelial cells and drives cellular senescence and brain microvascular dysfunction in a mouse model of tauopathy. Nat Commun. 2023;14:2367.
220. Gulej R, Nyúl-Tóth Á, Ahire C, et al. Elimination of senescent cells by treatment with Navitoclax/ABT263 reverses whole brain irradiation-induced blood-brain barrier disruption in the mouse brain. Geroscience. 2023;45:2983-3002.
221. Wang L, Wang B, Gasek NS, et al. Targeting p21Cip1 highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab. 2022;34:75-89.e8.
222. Lipskaia L, Breau M, Cayrou C, et al. mTert induction in p21-positive cells counteracts capillary rarefaction and pulmonary emphysema. EMBO Rep. 2024;25:1650-84.
223. Culley MK, Zhao J, Tai YY, et al. Frataxin deficiency promotes endothelial senescence in pulmonary hypertension. J Clin Invest. 2021;131:136459.
224. Gao Z, Santos RB, Rupert J, et al. Endothelial-specific telomerase inactivation causes telomere-independent cell senescence and multi-organ dysfunction characteristic of aging. Aging Cell. 2024;23:e14138.
225. Barinda AJ, Ikeda K, Nugroho DB, et al. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat Commun. 2020;11:481.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].