





Glycogen synthase kinase 3β: the nexus of chemoresistance, invasive capacity, and cancer stemness in pancreatic cancer

 , , ...
, , ... Abstract
The treatment of pancreatic cancer remains a significant clinical challenge due to the limited number of patients eligible for curative (R0) surgery, failures in the clinical development of targeted and immune therapies, and the pervasive acquisition of chemotherapeutic resistance. Refractory pancreatic cancer is typified by high invasiveness and resistance to therapy, with both attributes related to tumor cell stemness. These malignant characteristics mutually enhance each other, leading to rapid cancer progression. Over the past two decades, numerous studies have produced evidence of the pivotal role of glycogen synthase kinase (GSK)3β in the progression of over 25 different cancer types, including pancreatic cancer. In this review, we synthesize the current knowledge on the pathological roles of aberrant GSK3β in supporting tumor cell proliferation and invasion, as well as its contribution to gemcitabine resistance in pancreatic cancer. Importantly, we discuss the central role of GSK3β as a molecular hub that mechanistically connects chemoresistance, tumor cell invasion, and stemness in pancreatic cancer. We also discuss the involvement of GSK3β in the formation of desmoplastic tumor stroma and in promoting anti-cancer immune evasion, both of which constitute major obstacles to successful cancer treatment. Overall, GSK3β has characteristics of a promising therapeutic target to overcome chemoresistance in pancreatic cancer.

Keywords
INTRODUCTION
Approximately one-third of patients diagnosed with pancreatic ductal adenocarcinoma (PDAC) present with locally unresectable tumors, while nearly half exhibit distant metastatic tumors. Consequently, only 10%-15% of stage I or II patients can undergo resectable (R0) surgery, a significant proportion of whom eventually suffer local disease recurrence and/or distant metastasis post-surgery[1,2]. First-line chemotherapeutic regimens for palliative treatment of locally advanced pancreatic cancer and metastatic cases include FOLFIRINOX [a combination of folate, 5-fluorouracil (FU), irinotecan, and oxaliplatin][3], and a combination of nanoparticle albumin-bound (nab)-paclitaxel and gemcitabine[4]. The second-line regimen is FOLFIRI[5]. These multi-agent chemotherapy regimens offer only a modestly improved efficacy compared to gemcitabine monotherapy, and are appropriate only for a small proportion of pancreatic cancer patients with a good performance status (PS 0 or 1)[3-6]. Patients with a PS of 2 or higher, who represent most pancreatic cancer cases, undergo gemcitabine monotherapy[7,8], with many quickly developing resistance to the drug[9-11]. Unlike lung and colorectal cancer, neither preclinical studies nor clinical trials have yet to demonstrate significant efficacy against pancreatic cancer of rationally targeted agents, precision medicines based on the identification of actionable proto-oncoproteins, or immune checkpoint blockade[12-20]. Given these circumstances, the development of biology-based strategies against chemoresistance, specifically the resistance to gemcitabine by pancreatic cancer, is a pressing need in pancreatic cancer research[21].
MECHANISMS OF GEMCITABINE RESISTANCE IN PANCREATIC CANCER
Many reviews[9-11,22-25] have thoroughly examined the mechanisms of action of, and resistance to, gemcitabine in pancreatic cancer cells, detailing the steps of intra- and extracellular drug transportation[26-29], metabolic activation of gemcitabine[30-35], molecular pathways that combat drug-induced apoptosis[36-41], pro-oncogenic pathways that enable tumor cells to survive cytotoxic effects[36,42-51], and epithelial-mesenchymal transition (EMT), a pro-invasive tumor property[52,53]. These reviews[10,11,22-25] also discuss the role of specific
Efforts have been made to address resistance mechanisms that interfere with gemcitabine uptake and activation, with several compounds developed to enhance nucleoside transporter (NP) expression or bypass NPs and deoxycytidine kinase (dCK) through different mechanisms or chemical modifications of gemcitabine[9,11,64]. Several experimental studies have sought to reverse chemoresistance by activating multiple cell death pathways[65], deconstructing the desmoplastic stroma, and targeting immunosuppressive pathways within the hostile TME in pancreatic cancer[66-74]. Despite a decade of extensive research efforts, there has been limited therapeutic advancement in preclinical or clinical settings. This reaffirms the urgent need to identify mechanism-based strategies and therapeutic targets to overcome gemcitabine resistance in pancreatic cancer research.
MUTUAL DEPENDENCY OF CHEMORESISTANCE, TUMOR CELL INVASION, AND STEMNESS IN PANCREATIC CANCER
One of the fundamental challenges in addressing cancer drug resistance stems from the complex biological properties that tumor cells inherently possess (e.g., intrinsic resistance) or acquire during exposure to therapeutic agents (acquired resistance)[75]. Recent comprehensive reviews have detailed these properties, which include altered pharmacological reactions of tumor cells to drugs and their intermediate metabolites, clonal evolution of tumor cells to generate resistant clones, the latency and plasticity of cancer stem cells or tumor cells’ resilience to acquiring stemness phenotype, genetic and biological heterogeneity of tumor cells, activation of pro-survival pathways, impairment in cell death pathways, and adaptation to therapeutic pressures[76,77]. The aforementioned properties synergistically lead to increased resistance to therapy in various cancer types[77].
The concept of cancer stem cells (CSCs) has been proposed based on the observation that the uncontrolled proliferation of cancer is driven by a biologically distinct subset of tumor cells that are present in a relatively small proportion within the whole tumor[78,79]. CSCs are characterized by their ability to self-renew, sustain tumor propagation, express specific cell surface markers, and use multidrug efflux pumps. The clinical implication of CSCs lies in their potential to drive tumor regrowth after seemingly successful treatment with chemotherapeutics, radiation, or targeted agents because of their inherent resistance to therapy[80-83]. This has positioned CSCs as a critical therapeutic target, albeit one that is currently elusive due to their complex biological nature in intractable cancer types, including pancreatic cancer[84-90]. Several studies have shown that cancer therapy spares not only CSCs but also some residual cancer cells that acquire a CSC-like phenotype without mutation-based clonal selection, thus becoming resistant to therapy. This phenotypic switch is often associated with the activation of pro-oncogenic pathways such as Wnt- and Notch-mediated pathways[91]. Consistently, gemcitabine treatment can induce a shift towards a cancer stemness phenotype, primarily in gemcitabine-naïve pancreatic cancer cells[53,92-95] [Table 1, Figure 1].

Figure 1. The mechanistic interconnection of chemoresistance, tumor invasion, and cancer stemness presents in chemoresistant pancreatic cancer. Mechanisms responsible for the respective connections are described in Table 1. NF-κB: Nuclear factor-κB; ZEB1: zinc-finger-enhancer binding protein 1; EVA1: epithelial V-like antigen 1; MAL2: myelin and lymphocyte protein 2; NPs: nucleoside transporters; CD: cluster of differentiation; Oct4: octamer-binding transcription factor 4; NOx: nitrogen oxides; ROS: reactive oxygen species; STAT3: signal transducer and activator of transcription 3; ESA: epithelial-specific antigen; PDAC: pancreatic ductal adenocarcinoma.
Tumor stemness and pro-invasive properties of PDAC cells surviving the insult from continuous or repeated exposure to gemcitabine
| Mechanisms for gemcitabine-induced phenotypes | Ref. | |
| Cancer stemness phenotype | Acquisition of resistance to gemcitabine imparts stemness phenotype to the PDAC cells via the phosphorylation-mediated activation of c-Met receptor protein tyrosine kinase and the increased expression of CSC markers CD24 and CD44 | [53] |
| PDAC cells surviving gemcitabine treatment express the CSC markers: CD44, CD24, Oct4, and CD133, and an EMT marker slug | [92] | |
| Gemcitabine-resistant PDAC cells acquire cancer stemness and EMT phenotypes mediated by the NF-κB pathway | [93] | |
| Gemcitabine treatment promotes chemoresistance and cancer stemness through the NOx/ROS/NF-κB/STAT3 signaling cascade | [94] | |
| Gemcitabine-resistant PDAC cells show the activation of c-Met receptor protein tyrosine kinase and the increased expression of CSC markers CD24, CD44, and ESA | [95] | |
| Pro-invasive phenotype | Gemcitabine-resistant PDAC cells show spindle shape with pseudopodia, increased expression of vimentin, and decreased expression of E-cadherin | [53] |
| Gemcitabine-resistant PDAC cells acquire EMT phenotype by activation of the Notch signaling pathway | [49] | |
| Gemcitabine-resistant PDAC cells show the gene expression profile responsible for EMT phenotype | [115] | |
| PDAC cells surviving gemcitabine treatment express the CSC markers: CD44, CD24, Oct4, and CD133, and an EMT marker slug | [92] | |
| Gemcitabine-resistant PDAC cells acquire cancer stemness and EMT phenotypes mediated by the NF-κB pathway | [93] | |
| Gemcitabine-resistant PDAC cells showed increased expression of ZEB1 | [52] | |
| Suppression of EMT leads to an increase in PDAC cell proliferation with enhanced expression of NPs in tumors, contributing to enhanced sensitivity to gemcitabine and increased survival of mouse models of PDAC with deletion of snail or twist | [11] | |
| Gemcitabine-resistant PDAC cells show spindle shape with pseudopodia, migratory and invasive capacity, increased vimentin and decreased E-cadherin expression, and nuclear localization of β-catenin | [95] |
Highly invasive capacity and therapy resistance are the defining biological and clinical characteristics of pancreatic cancer, often resulting in treatment failure and poor patient outcomes[1,2]. Although not sufficient on its own, tumor invasion serves as an initial step in the complex process leading to cancer progression and metastasis[96]. EMT, which modifies the morphological and functional behaviors of cancer cells to resemble mesenchymal cell types, is a key prerequisite for the invasion seen in many cancer types[97,98], including pancreatic cancer[99]. A growing body of research has demonstrated the causal link between chemotherapeutic stimuli and EMT in various cancer types, such as colon[100,101], ovarian[102], breast[103-107], and skin squamous cell carcinoma[108]. These studies have given rise to a concept that chemoresistance and the tumor invasion-metastasis cascade are interrelated processes that accelerate cancer progression[109-112]. In line with this concept and the paradox of treatment-induced metastasis, suggesting that nearly all cancer treatments can inadvertently trigger and facilitate metastatic spread[113,114], several studies have reported an association of resistance to gemcitabine with EMT and invasion in pancreatic cancer[49,52,53,92,93,95,115,116]
Regardless of therapy resistance, mounting evidence has shown a strong association between the pro-invasive phenotype (represented by EMT) and CSCs in many cancer types[117-120], including pancreatic cancer[121], because of shared distinct biological mechanisms. Consistent with this connection, many of the studies referenced in Table 1 have reported a simultaneous association of resistance to gemcitabine with both pro-invasive and cancer stemness phenotypes[52,53,92,93,95]. This highlights a three-way (or triangular) interaction among chemoresistance, tumor invasion, and cancer stemness in pancreatic cancer [Figure 1], as previously suggested[122,123]. We have established gemcitabine-sensitive pancreatic cancer BxPC-3 derivative cell clones that gained stepwise resistance to gemcitabine (BxG30, BxG140, and BxG400 in increasing order of resistance)[124]. We have recently observed an increased invasive capacity with the formation of characteristic cellular surface microstructures (lamellipodia[125] and invadopodia[126]), and sphere-forming ability in the resistant cell clones compared to their parental BxPC-3 cells [Figure 2A]. Moreover, we have observed severe local tumor invasion and metastasis to the liver and peritoneum in mice orthotopically transplanted with the most resistant BxG400 cells [Figure 2B], which models the refractory pancreatic cancer patients developing resistance to gemcitabine. Our preliminary observations reaffirm the interdependency between these malignant properties in pancreatic cancer cells acquiring resistance to gemcitabine.
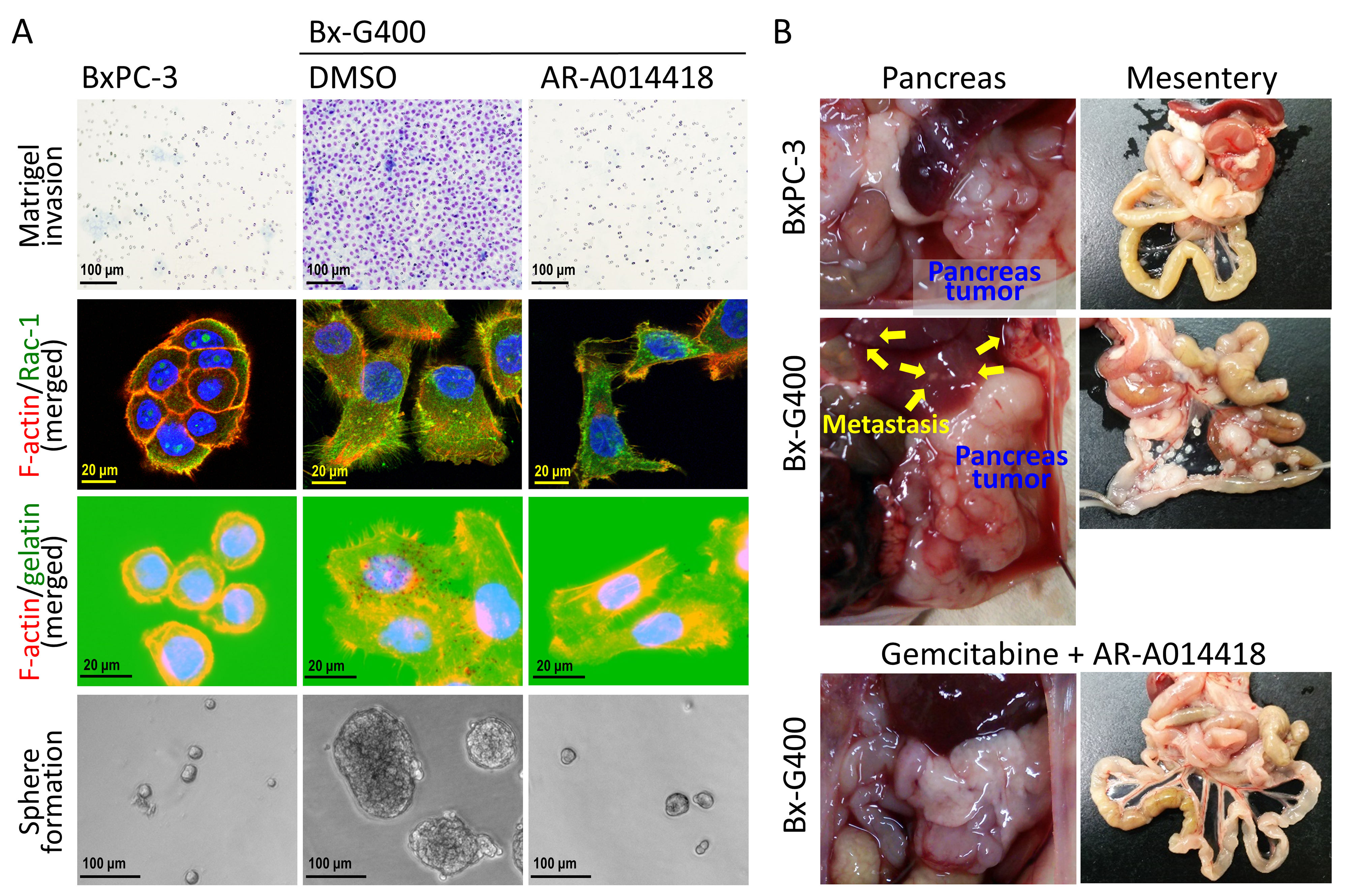
Figure 2. (A) Representative findings of matrigel invasion (upper panels), formation of lamellipodia and invadopodia [middle six panels: nuclei were counterstained with DAPI (blue fluorescence), and sphere formation (lower panels) of BxPC-3 cells and BxPC-3 cell-derived gemcitabine-resistant Bx-G400 cells that were treated with DMSO (diluent of the inhibitor) and GSK3β inhibitor, AR-A014418; (B) Laparotomy findings of the mice with intrapancreatic transplantation of BxPC-3 and Bx-G400 cells, respectively. The bottom panels show the mice treated with gemcitabine and AR-A014418 in combination for 8 weeks after transplantation. DAPI: 4’,6-diamidino-2-phenylindole; DMSO: dimethyl sulfoxide; GSK3β: glycogen synthase kinase 3β.
OUTLINE OF GLYCOGEN SYNTHASE KINASE 3β BIOLOGY AND ITS INVOLVEMENT IN DISEASES
Glycogen synthase kinase (GSK)3β is an isoform of the GSK3 family of serine (S)/threonine (T) protein kinases. GSK3β is involved in a multitude of biological processes and pathways in the complex molecular networks of cells, where it interacts with nearly 100 or more structural and functional proteins via phosphorylation[127,128]. The enzymatic activity of GSK3β is regulated by the differential phosphorylation of its S9 (inactive form) and tyrosine (Y)216 (active form) residues. Even though GSK3β is typically active in cells, inhibitory regulation of its activity primarily benefits normal cells in maintaining their vital activity and homeostasis in response to various extracellular and intracellular stimuli[127-129]. Due to the redundancy in cellular expression and functions between GSK3β and its isoform GSK3α, the pathological roles of GSK3β have garnered increased attention. Aberrant activity and expression of GSK3β, as well as defects in its inhibitory regulation, contribute to the pathogenesis and progression of common diseases, including type 2 diabetes mellitus, neurodegenerative diseases (e.g., Alzheimer’s disease), inflammatory and immunological diseases, and cancer[130,131]. Given its counteracting functions in normal cells and diseases, GSK3β is considered a potential therapeutic target in major health disorders, thereby driving the identification and development of pharmacological GSK3β inhibitors[132-136].
The biochemistry, biology, and functions of GSK3 family kinases (GSK3α and GSK3β) in normal cells, as well as their involvement in a wide range of common diseases, have increasingly attracted scientific attention in biomedical and pharmacological fields. This topic has been extensively reviewed in previous literature[127-136], and is therefore only briefly outlined here.
TUMOR-PROMOTING ROLES OF GSK3β IN PANCREATIC CANCER
Contrary to its pathological roles in diseases other than cancer, activated GSK3β counters pro-oncogenic pathways such as those mediated by Wnt/β-catenin, Hedgehog, and Notch signaling and transcription factors (e.g., snail) that induce EMT in normal cells. Given its functions in non-transformed cells, activation of GSK3β has long been hypothesized to suppress the development and progression of cancer[137-139], thereby posing a challenge to pharmaceutical industries and clinical oncologists aiming to develop and apply GSK3β inhibitors for cancer treatment. However, no direct evidence supports the tumor-suppressor role of this kinase, nor the effect of GSK3β inhibition on promoting cancer development and progression. Contrary to this hypothesis, a significant amount of research conducted by our group and others over the last two decades has provided solid evidence demonstrating the tumor-promoting roles of active GSK3β as well as therapeutic effects related to its inhibition in more than 25 different cancer types (reviewed in[140-146]), including pancreatic cancer (reviewed in[147-152]). As a result, GSK3β has emerged as a potential therapeutic target in cancer, encouraging the development of GSK3β inhibitors for cancer treatment[153-155]. Similar to other cancer types, accumulating evidence for pancreatic cancer[156-182] shows that deregulated GSK3β supports tumor cell survival, immortality, and proliferation by mediating distinct pathways. As discussed below, GSK3β also facilitates the invasion of tumor cells and makes them unresponsive or resistant to chemotherapy and ionizing radiation [Figure 3].

Figure 3. Tumor-promoting properties of GSK3β and underlying mechanisms reported in pancreatic cancer[156-182]. Dotted arrow: preliminary findings (by Domoto et al.). The mechanisms responsible for the respective interconnections indicated by red bidirectional arrows are shown in Table 1 and Figure 1. GSK3β: Glycogen synthase kinase 3β; NF-κB: nuclear factor κB; Bcl-2: B-cell/chronic lymphocytic leukemia lymphoma 2; hTERT: human telomerase reverse transcriptase; XIAP: X-linked inhibitor of apoptosis protein; TRAIL: tumor necrosis factor-related apoptosis-inducing ligand; JNK: c-Jun N-terminal kinase; TFEB: transcription factor EB; STAT3: signal transducer and activator of transcription 3; TP53INP1: tumor protein 53-inducible nuclear protein 1; E2F1: E2 transcription factor 1; TopBP1: topoisomerase II binding protein 1; ATR: ataxia telangiectasia and Rad3-related; Chk1: checkpoint kinase 1; FAK: focal adhesion kinase; CXCR4: C-X-C chemokine receptor type 4; MMP: matrix metalloproteinase; CXCL: C-X-C chemokine ligand; SOX2: sex-determining region Y-box transcription factor 2.
GSK3β AS A MOLECULAR HUB IN MECHANISTICALLY WIRING THE CHEMORESISTANCE, TUMOR INVASION AND CANCER STEMNESS
As depicted in Figure 3, most studies on pancreatic cancer have demonstrated that the deregulated GSK3β sustains tumor cell survival, immortalization, and proliferation - common and fundamental features that engender therapy resistance in nearly all cancer types. This is achieved by enhancing cell immortality and several pro-oncogenic pathways [e.g., Nuclear factor-κB (NF-κB), Notch, K-ras, c-Myc], and by abrogating distinct tumor suppressor pathways (e.g., Rb) [Figure 3]. Our group and others have previously reported that GSK3β contributes to the unresponsiveness of pancreatic cancer cells to gemcitabine by impairing the DNA damage response mediated by tumor protein 53-inducible nuclear protein 1 (TP53INP1) and topoisomerase IIβ binding protein 1 (TopBP1)/ataxia telangiectasia and Rad3-related (ATR)/checkpoint kinase 1 (Chk1)[165,169,178]. We have also shown that GSK3β plays a role in the acquisition of resistance to gemcitabine in resistant pancreatic cancer cell clones derived from BxPC-3[124] [Figure 2], via impairing the functional interaction between the Rb tumor suppressor protein and pro-oncogenic E2 transcription factor (E2F)1[179]. Our previous studies on gemcitabine-unresponsive pancreatic cancer and temozolomide-resistant glioblastoma cells have indicated that GSK3β enhances tumor cell migration and invasion via the focal adhesion kinase (FAK), Rac1, and c-Jun N-terminal kinase (JNK)-mediated pathway[169,183]. It is conceivable that the respective mechanisms responsible for chemoresistance and tumor invasion share GSK3β as a common trigger for both malignant properties in gemcitabine-resistant pancreatic cancer.
As we previously reviewed[146], a series of studies have shown that GSK3β underpins the pivotal mechanisms for sustaining CSCs and the acquisition of cancer stemness phenotype in various cancer types, including colorectal cancer, prostate cancer, head and neck squamous cell carcinoma, glioblastoma, and leukemia. We have previously shown that kenpaullone, an adenosine triphosphate (ATP)-competitive GSK3β inhibitor, reversed the temozolomide resistance of glioblastoma patient-derived tumor stem cells[184]. In a recent preliminary study, we found that GSK3β inhibition counteracts tumor invasion and distant metastasis
PRESUMPTIVE INVOLVEMENT OF GSK3β IN THE DESMOPLASTIC TUMOR STROMA AND THE PERMISSIVE ANTI-CANCER IMMUNE ENVIRONMENT
Desmoplastic tumor stroma and permissive (or tolerant) immunity against cancer make up the hostile TME[60-62] that is recognized as a formidable obstacle for palliative treatment with chemotherapeutics, radiation, and targeted agents in pancreatic cancer[70-73]. Here, we briefly discuss the prospective involvement of GSK3β in the pancreatic cancer TME.
The primary cellular components of pancreatic cancer TME include pancreatic stellate cells (PSCs)[185] and cancer-associated fibroblasts (CAFs)[60,186,187]. Both stromal cell types in pancreatic cancer support tumor cells and produce dense fibrous stroma that forms a physical barrier for drug delivery, and their interaction with tumor cells mechanistically contributes to chemoresistance. PSCs, a minor and quiescent cellular population in healthy pancreas stroma, are activated by extracellular stimulants including tumor necrosis factor
The permissive immune environment in pancreatic cancer is complex, primarily resulting from the failure in innate immunity exerted by natural killer (NK) T-cells and the suppression of adaptive immunity by the immune checkpoint machinery between CD8+ T-cells and tumor cells, mediated by the programmed death (PD)-1/PD ligand (PD-L)1 axis and cytotoxic T-lymphocyte-associated protein (CTLA)-4. Despite substantial and excellent preclinical backing, most clinical studies have failed to prove the efficacy of antibody-based immune checkpoint blockades (reviewed in[18,19,199-202]). We[146] and a recent series of reviews[151,154,155] have detailed the reported evidence showing that GSK3β attenuates the ability of anti-tumor immunocellular arsenals such as NK cells, CD8+ T-cell-derived pluripotent memory stem T-cells with cytotoxic capacity, and tumor-type specific genetically engineered chimeric antigen receptor (CAR)-T cells. The reported evidence also showed that GSK3β enhances PD-1 expression depending on the transcription factor TBX21 (Tbet) and PD-L1 expression in response to the inhibition of poly [ADP-ribose] polymerase (PARP)1, and that inhibition of GSK3β reverses the blockade of CD28’s ability to bind and stimulate antigen-presenting immune cells by CTLA-4. Importantly, a recent study demonstrated that FAK suppresses antigen processing and presentation to promote immune evasion in pancreatic cancer[203,204], suggesting a previously underexplored mechanistic link between desmoplastic tumor stroma and tumor cell-autonomous mechanisms of immune evasion[205-207]. If future studies provide a direct relationship between tolerant anti-tumor immunity and the acquisition of chemoresistance, it will enhance the opportunity to combat chemoresistance in pancreatic cancer by targeting GSK3β.
CONCLUSION
GSK3β potentially functions as a molecular hub that wires chemoresistance, tumor invasion, and cancer stemness phenotype, thereby aggravating pancreatic cancer towards the incurable/devastating disease stage.
DECLARATIONS
Authors’ contributions
Conceptualization: Minamoto T
Literature search: Uehara M, Minamoto T
Preparation of figures: Domoto T, Minamoto T
Original drafting: Uehara M, Domoto T, Minamoto T
Review and editing: Takenaka S, Takeuchi O, Shimasaki T, Miyashita T, Minamoto T
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, Technology and Culture and from the Japan Society for the Promotion of Science (to UM: 23K14644; TD: 22K07227, T. Miyashita: 23K08163; and T. Minamoto: 22H03144).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
Supplementary Materials
REFERENCES
3. Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801-10.
4. Goldstein D, El-Maraghi RH, Hammel P, et al. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial. J Natl Cancer Inst 2015;107:dju413.
5. Wang-Gillam A, Li CP, Bodoky G, et al; NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545-57.
6. Tempero MA, Malafa MP, Chiorean EG, et al. NCCN guidelines insights: pancreatic adenocarcinoma, version 1.2019. J Natl Compr Canc Netw 2019;17:202-10.
7. Springfeld C, Jäger D, Büchler MW, et al. Chemotherapy for pancreatic cancer. Presse Med 2019;48:e159-74.
8. Christenson ES, Jaffee E, Azad NS. Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: a bright future. Lancet Oncol 2020;21:e135-45.
9. Bergman AM, Pinedo HM, Peters GJ. Determinants of resistance to 2',2'-difluorodeoxycytidine (gemcitabine). Drug Resist Updat 2002;5:19-33.
10. Binenbaum Y, Na’ara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist Updat 2015;23:55-68.
11. Zeng S, Pöttler M, Lan B, Grützmann R, Pilarsky C, Yang H. Chemoresistance in pancreatic cancer. Int J Mol Sci 2019;20:4504.
12. Sun J, Russell CC, Scarlett CJ, McCluskey A. Small molecule inhibitors in pancreatic cancer. RSC Med Chem 2020;11:164-83.
13. Carbone D, Pecoraro C, Panzeca G, et al. 1,3,4-Oxadiazole and 1,3,4-thiadiazole nortopsentin derivatives against pancreatic ductal adenocarcinoma: synthesis, cytotoxic activity, and inhibition of CDK1. Mar Drugs 2023;21:412.
14. Pishvaian MJ, Blais EM, Brody JR, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol 2020;21:508-18.
15. Al-Share B, Hammad N, Diab M. Pancreatic adenocarcinoma: molecular drivers and the role of targeted therapy. Cancer Metastasis Rev 2021;40:355-71.
16. O’Kane GM, Lowery MA. Moving the needle on precision medicine in pancreatic cancer. J Clin Oncol 2022;40:2693-705.
17. Jiang H, Hegde S, Knolhoff BL, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 2016;22:851-60.
18. Balachandran VP, Beatty GL, Dougan SK. Broadening the impact of immunotherapy to pancreatic cancer: challenges and opportunities. Gastroenterology 2019;156:2056-72.
19. Ullman NA, Burchard PR, Dunne RF, Linehan DC. Immunologic strategies in pancreatic cancer: making cold tumors hot. J Clin Oncol 2022;40:2789-805.
20. Hosein AN, Dougan SK, Aguirre AJ, Maitra A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat Cancer 2022;3:272-86.
21. Yang G, Guan W, Cao Z, et al. Integrative genomic analysis of gemcitabine resistance in pancreatic cancer by patient-derived xenograft models. Clin Cancer Res 2021;27:3383-96.
22. de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol 2014;741:8-16.
23. Jia Y, Xie J. Promising molecular mechanisms responsible for gemcitabine resistance in cancer. Genes Dis 2015;2:299-306.
24. Rajabpour A, Rajaei F, Teimoori-Toolabi L. Molecular alterations contributing to pancreatic cancer chemoresistance. Pancreatology 2017;17:310-20.
25. Adamska A, Elaskalani O, Emmanouilidi A, et al. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv Biol Regul 2018;68:77-87.
26. Rauchwerger DR, Firby PS, Hedley DW, Moore MJ. Equilibrative-sensitive nucleoside transporter and its role in gemcitabine sensitivity. Cancer Res 2000;60:6075-9.
27. Nakano Y, Tanno S, Koizumi K, et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer 2007;96:457-63.
28. Hagmann W, Jesnowski R, Löhr JM. Interdependence of gemcitabine treatment, transporter expression, and resistance in human pancreatic carcinoma cells. Neoplasia 2010;12:740-7.
29. Hagmann W, Faissner R, Schnölzer M, Löhr M, Jesnowski R. Membrane drug transporters and chemoresistance in human pancreatic carcinoma. Cancers 2010;3:106-25.
30. Saiki Y, Yoshino Y, Fujimura H, et al. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem Biophys Res Commun 2012;421:98-104.
31. Dash S, Ueda T, Komuro A, et al. MYC/glutamine dependency is a therapeutic vulnerability in pancreatic cancer with deoxycytidine kinase inactivation-induced gemcitabine resistance. Mol Cancer Res 2023;21:444-57.
32. Costantino CL, Witkiewicz AK, Kuwano Y, et al. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res 2009;69:4567-72.
33. Nakahira S, Nakamori S, Tsujie M, et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int J Cancer 2007;120:1355-63.
34. Wang C, Zhang W, Fu M, Yang A, Huang H, Xie J. Establishment of human pancreatic cancer gemcitabine-resistant cell line with ribonucleotide reductase overexpression. Oncol Rep 2015;33:383-90.
35. Minami K, Shinsato Y, Yamamoto M, et al. Ribonucleotide reductase is an effective target to overcome gemcitabine resistance in gemcitabine-resistant pancreatic cancer cells with dual resistant factors. J Pharmacol Sci 2015;127:319-25.
36. Ng SSW, Tsao MS, Chow S, Hedley DW. Inhibition of phosphatidylinositide 3-kinase enhances gemcitabine-induced apoptosis in human pancreatic cancer cells. Cancer Res 2000;60:5451-5.
37. Akada M, Crnogorac-Jurcevic T, Lattimore S, et al. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin Cancer Res 2005;11:3094-101.
38. Duxbury MS, Ito H, Benoit E, Waseem T, Ashley SW, Whang EE. RNA interference demonstrates a novel role for integrin-linked kinase as a determinant of pancreatic adenocarcinoma cell gemcitabine chemoresistance. Clin Cancer Res 2005;11:3433-8.
39. Bafna S, Kaur S, Momi N, Batra SK. Pancreatic cancer cells resistance to gemcitabine: the role of MUC4 mucin. Br J Cancer 2009;101:1155-61.
40. Duxbury MS, Ito H, Benoit E, Waseem T, Ashley SW, Whang EE. A novel role for carcinoembryonic antigen-related cell adhesion molecule 6 as a determinant of gemcitabine chemoresistance in pancreatic adenocarcinoma cells. Cancer Res 2004;64:3987-93.
41. Giroux V, Malicet C, Barthet M, et al. p8 is a new target of gemcitabine in pancreatic cancer cells. Clin Cancer Res 2006;12:235-41.
42. Bhardwaj V, Tadinada SM, Lai JCK, Bhushan A. Failure of pancreatic cancer chemotherapy: consequences of drug resistance mechanisms. In: Srivastava SK, editor. Pancreatic cancer - molecular mechanism and targets. InTech; 2012. Available from: https://www.intechopen.com/chapters/33494. [Last accessed on 29 Jan 2024].
43. Wey JS, Gray MJ, Fan F, et al. Overexpression of neuropilin-1 promotes constitutive MAPK signalling and chemoresistance in pancreatic cancer cells. Br J Cancer 2005;93:233-41.
44. Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Inhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells. Clin Cancer Res 2004;10:2307-18.
45. Arlt A, Gehrz A, Müerköster S, et al. Role of NF-κB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 2003;22:3243-51.
46. Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-κB-regulated gene products. Cancer Res 2007;67:3853-61.
47. Pan X, Arumugam T, Yamamoto T, et al. Nuclear factor-κB p65/relA silencing induces apoptosis and increases gemcitabine effectiveness in a subset of pancreatic cancer cells. Clin Cancer Res 2008;14:8143-51.
48. Singh S, Srivastava SK, Bhardwaj A, Owen LB, Singh AP. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer 2010;103:1671-9.
49. Wang Z, Li Y, Kong D, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 2009;69:2400-7.
50. Yao J, Qian C. Inhibition of Notch3 enhances sensitivity to gemcitabine in pancreatic cancer through an inactivation of PI3K/Akt-dependent pathway. Med Oncol 2010;27:1017-22.
51. Yao J, An Y, Wie JS, et al. Cyclopamine reverts acquired chemoresistance and down-regulates cancer stem cell markers in pancreatic cancer cell lines. Swiss Med Wkly 2011;141:w13208.
52. Meidhof S, Brabletz S, Lehmann W, et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med 2015;7:831-47.
53. Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol 2007;14:3629-37.
54. Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br J Cancer 2013;108:1-8.
55. Hamada S, Masamune A, Shimosegawa T. Inflammation and pancreatic cancer: disease promoter and new therapeutic target. J Gastroenterol 2014;49:605-17.
56. Bijlsma MF, van Laarhoven HWM. The conflicting roles of tumor stroma in pancreatic cancer and their contribution to the failure of clinical trials: a systematic review and critical appraisal. Cancer Metastasis Rev 2015;34:97-114.
57. Neesse A, Algül H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut 2015;64:1476-84.
58. Rath N, Olson MF. Regulation of pancreatic cancer aggressiveness by stromal stiffening. Nat Med 2016;22:462-3.
59. DuFort CC, DelGiorno KE, Hingorani SR. Mounting pressure in the microenvironment: fluids, solids, and cells in pancreatic ductal adenocarcinoma. Gastroenterology 2016;150:1545-57.e2.
60. Whittle MC, Hingorani SR. Fibroblasts in pancreatic ductal adenocarcinoma: biological mechanisms and therapeutic targets. Gastroenterology 2019;156:2085-96.
61. Herting CJ, Karpovsky I, Lesinski GB. The tumor microenvironment in pancreatic ductal adenocarcinoma: current perspectives and future directions. Cancer Metastasis Rev 2021;40:675-89.
62. Hingorani SR. Epithelial and stromal co-evolution and complicity in pancreatic cancer. Nat Rev Cancer 2023;23:57-77.
63. Geller LT, Barzily-Rokni M, Danino T, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017;357:1156-60.
64. Saiki Y, Hirota S, Horii A. Attempts to remodel the pathways of gemcitabine metabolism: recent approaches to overcoming tumours with acquired chemoresistance. Cancer Drug Resist 2020;3:819-31.
65. Santofimia-Castaño P, Iovanna J. Combating pancreatic cancer chemoresistance by triggering multiple cell death pathways. Pancreatology 2021;21:522-9.
66. Garber K. Stromal depletion goes on trial in pancreatic cancer. J Natl Cancer Inst 2010;102:448-50.
67. Erkan M, Hausmann S, Michalski CW, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol 2012;9:454-67.
68. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418-29.
69. Vennin C, Murphy KJ, Morton JP, Cox TR, Pajic M, Timpson P. Reshaping the tumor stroma for treatment of pancreatic cancer. Gastroenterology 2018;154:820-38.
70. Neesse A, Bauer CA, Öhlund D, et al. Stromal biology and therapy in pancreatic cancer: ready for clinical translation? Gut 2019;68:159-71.
71. Huang H, Brekken RA. The next wave of stroma-targeting therapy in pancreatic cancer. Cancer Res 2019;79:328-30.
72. Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol 2020;17:487-505.
73. Carpenter ES, Steele NG, Pasca di Magliano M. Targeting the microenvironment to overcome gemcitabine resistance in pancreatic cancer. Cancer Res 2020;80:3070-1.
74. Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat Rev Clin Oncol 2020;17:527-40.
75. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013;13:714-26.
77. Tyner JW, Haderk F, Kumaraswamy A, et al. Understanding drug sensitivity and tackling resistance in cancer. Cancer Res 2022;82:1448-60.
78. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer 2012;12:133-43.
81. Maugeri-Saccà M, Vigneri P, De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res 2011;17:4942-7.
82. Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol 2018;53:156-67.
83. Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer 2018;18:669-80.
84. Borah A, Raveendran S, Rochani A, Maekawa T, Kumar DS. Targeting self-renewal pathways in cancer stem cells: clinical implications for cancer therapy. Oncogenesis 2015;4:e177.
85. Kuhlmann JD, Hein L, Kurth I, Wimberger P, Dubrovska A. Targeting cancer stem cells: promises and challenges. Anticancer Agents Med Chem 2016;16:38-58.
86. Clarke MF. Clinical and therapeutic implications of cancer stem cells. N Engl J Med 2019;380:2237-45.
87. Raj D, Aicher A, Heeschen C. Concise review: stem cells in pancreatic cancer: from concept to translation. Stem Cells 2015;33:2893-902.
88. Sancho P, Alcala S, Usachov V, Hermann PC, Sainz B Jr. The ever-changing landscape of pancreatic cancer stem cells. Pancreatology 2016;16:489-96.
89. Hermann PC, Sainz B Jr. Pancreatic cancer stem cells: a state or an entity? Semin Cancer Biol 2018;53:223-31.
90. Patil K, Khan FB, Akhtar S, Ahmad A, Uddin S. The plasticity of pancreatic cancer stem cells: implications in therapeutic resistance. Cancer Metastasis Rev 2021;40:691-720.
91. Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: 'What does not kill me strengthens me'. Br J Cancer 2015;112:1725-32.
92. Quint K, Tonigold M, Di Fazio P, et al. Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epithelial-mesenchymal transition. Int J Oncol 2012;41:2093-102.
93. Zhang Y, Wei J, Wang H, et al. Epithelial mesenchymal transition correlates with CD24+CD44+ and CD133+ cells in pancreatic cancer. Oncol Rep 2012;27:1599-605.
94. Zhang Z, Duan Q, Zhao H, et al. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-κB/STAT3 signaling cascade. Cancer Lett 2016;382:53-63.
95. Kuo YC, Kou HW, Hsu CP, Lo CH, Hwang TL. Identification and clinical significance of pancreatic cancer stem cells and their chemotherapeutic drug resistance. Int J Mol Sci 2023;24:7331.
98. LeBleu VS, Thiery JP. The continuing search for causality between epithelial-to-mesenchymal transition and the metastatic fitness of carcinoma cells. Cancer Res 2022;82:1467-9.
99. Beuran M, Negoi I, Paun S, et al. The epithelial to mesenchymal transition in pancreatic cancer: a systematic review. Pancreatology 2015;15:217-25.
100. Yang AD, Fan F, Camp ER, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 2006;12:4147-53.
101. Dallas NA, Xia L, Fan F, et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res 2009;69:1951-7.
102. Kajiyama H, Shibata K, Terauchi M, et al. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 2007;31:277-83.
103. Hiscox S, Morgan L, Barrow D, Dutkowskil C, Wakeling A, Nicholson RI. Tamoxifen resistance in breast cancer cells is accompanied by an enhanced motile and invasive phenotype: inhibition by gefitinib ('Iressa', ZD1839). Clin Exp Metastasis 2004;21:201-12.
104. Hiscox S, Jiang WG, Obermeier K, et al. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of β-catenin phosphorylation. Int J Cancer 2006;118:290-301.
105. Li QQ, Xu JD, Wang WJ, et al. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res 2009;15:2657-65.
106. Fischer KR, Durrans A, Lee S, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015;527:472-6.
107. Fatherree JP, Guarin JR, McGinn RA, Naber SP, Oudin MJ. Chemotherapy-induced collagen IV drives cancer cell motility through activation of src and focal adhesion kinase. Cancer Res 2022;82:2031-44.
108. Debaugnies M, Rodríguez-Acebes S, Blondeau J, et al. RHOJ controls EMT-associated resistance to chemotherapy. Nature 2023;616:168-75.
109. Alexander S, Friedl P. Cancer invasion and resistance: interconnected processes of disease progression and therapy failure. Trends Mol Med 2012;18:13-26.
110. van Staalduinen J, Baker D, ten Dijke P, van Dam H. Epithelial-mesenchymal-transition-inducing transcription factors: new targets for tackling chemoresistance in cancer? Oncogene 2018;37:6195-211.
111. Garg M. Emerging roles of epithelial-mesenchymal plasticity in invasion-metastasis cascade and therapy resistance. Cancer Metastasis Rev 2022;41:131-45.
112. Weiss F, Lauffenburger D, Friedl P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat Rev Cancer 2022;22:157-73.
113. Ebos JML. Prodding the beast: assessing the impact of treatment-induced metastasis. Cancer Res 2015;75:3427-35.
114. Karagiannis GS, Condeelis JS, Oktay MH. Chemotherapy-induced metastasis: molecular mechanisms, clinical manifestations, therapeutic interventions. Cancer Res 2019;79:4567-76.
115. Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res 2009;69:5820-8.
116. Zheng X, Carstens JL, Kim J, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015;527:525-30.
117. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009;9:265-73.
118. Sato R, Semba T, Saya H, Arima Y. Concise review: stem cells and epithelial-mesenchymal transition in cancer: biological implications and therapeutic targets. Stem Cells 2016;34:1997-2007.
119. Zheng X, Dai F, Feng L, Zou H, Feng L, Xu M. Communication between epithelial-mesenchymal plasticity and cancer stem cells: new insights into cancer progression. Front Oncol 2021;11:617597.
120. Lambert AW, Weinberg RA. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer 2021;21:325-38.
121. Ishiwata T. Cancer stem cells and epithelial-mesenchymal transition: novel therapeutic targets for cancer. Pathol Int 2016;66:601-8.
122. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010;29:4741-51.
123. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 2017;14:611-29.
124. Yoneyama H, Takizawa-Hashimoto A, Takeuchi O, et al. Acquired resistance to gemcitabine and cross-resistance in human pancreatic cancer clones. Anticancer Drugs 2015;26:90-100.
125. Machesky LM. Lamellipodia and filopodia in metastasis and invasion. FEBS Lett 2008;582:2102-11.
126. Paz H, Pathak N, Yang J. Invading one step at a time: the role of invadopodia in tumor metastasis. Oncogene 2014;33:4193-202.
127. Cormier KW, Woodgett JR. Recent advances in understanding the cellular roles of GSK-3. F1000Res 2017;6:167.
128. Patel P, Woodgett JR. Chapter eight - Glycogen synthase kinase 3: a kinase for all pathways? Curr Top Dev Biol 2017;123:277-302.
130. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 2015;148:114-31.
131. Hoffmeister L, Diekmann M, Brand K, Huber R. GSK3: a kinase balancing promotion and resolution of inflammation. Cells 2020;9:820.
132. Pandey MK, DeGrado TR. Glycogen synthase kinase-3 (GSK-3)-targeted therapy and imaging. Theranostics 2016;6:571-93.
133. Khan I, Tantray MA, Alam MS, Hamid H. Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur J Med Chem 2017;125:464-77.
134. Palomo V, Martinez A. Glycogen synthase kinase 3 (GSK-3) inhibitors: a patent update (2014-2015). Expert Opin Ther Pat 2017;27:657-66.
135. Saraswati AP, Ali Hussaini SM, Krishna NH, Babu BN, Kamal A. Glycogen synthase kinase-3 and its inhibitors: potential target for various therapeutic conditions. Eur J Med Chem 2018;144:843-58.
136. Wei J, Wang J, Zhang J, Yang J, Wang G, Wang Y. Development of inhibitors targeting glycogen synthase kinase-3β for human diseases: strategies to improve selectivity. Eur J Med Chem 2022;236:114301.
137. Luo J. Glycogen synthase kinase 3β (GSK3β) in tumorigenesis and cancer chemotherapy. Cancer Lett 2009;273:194-200.
138. McCubrey JA, Davis NM, Abrams SL, et al. Diverse roles of GSK-3: tumor promoter-tumor suppressor, target in cancer therapy. Adv Biol Regul 2014;54:176-96.
139. Tejeda-Muñoz N, Robles-Flores M. Glycogen synthase kinase 3 in Wnt signaling pathway and cancer. IUBMB Life 2015;67:914-22.
140. Miyashita K, Nakada M, Shakoori A, et al. An emerging strategy for cancer treatment targeting aberrant glycogen synthase kinase 3β. Anticancer Agents Med Chem 2009;9:1114-22.
141. McCubrey JA, Steelman LS, Bertrand FE, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014;5:2881-911.
142. Domoto T, Pyko IV, Furuta T, et al. Glycogen synthase kinase-3β is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci 2016;107:1363-72.
143. Walz A, Ugolkov A, Chandra S, et al. Molecular pathways: revisiting glycogen synthase kinase-3β as a target for the treatment of cancer. Clin Cancer Res 2017;23:1891-7.
144. Nagini S, Sophia J, Mishra R. Glycogen synthase kinases: moonlighting proteins with theranostic potential in cancer. Semin Cancer Biol 2019;56:25-36.
145. Duda P, Akula SM, Abrams SL, et al. Targeting GSK3 and associated signaling pathways involved in cancer. Cells 2020;9:1110.
146. Domoto T, Uehara M, Bolidong D, Minamoto T. Glycogen synthase kinase 3β in cancer biology and treatment. Cells 2020;9:1388.
147. Garcea G, Manson MM, Neal CP, et al. Glycogen synthase kinase-3 beta; a new target in pancreatic cancer? Curr Cancer Drug Targets 2007;7:209-15.
148. Shimasaki T, Kitano A, Motoo Y, Minamoto T. Aberrant glycogen synthase kinase 3β in the development of pancreatic cancer. J Carcinog 2012;11:15.
149. Zhang Q, Bhojani MS, Ben-Josef E, et al. Glycogen synthase kinase 3β in pancreatic cancer and its implications in chemotherapy and radiation therapy. J Carcinog Mutagen 2013;4:147.
150. Pecoraro C, Faggion B, Balboni B, et al. GSK3β as a novel promising target to overcome chemoresistance in pancreatic cancer. Drug Resist Updat 2021;58:100779.
151. Park R, Coveler AL, Cavalcante L, Saeed A. GSK-3β in pancreatic cancer: spotlight on 9-ING-41, its therapeutic potential and immune modulatory properties. Biology 2021;10:610.
152. Elmadbouh OHM, Pandol SJ, Edderkaoui M. Glycogen synthase kinase 3β: a true foe in pancreatic cancer. Int J Mol Sci 2022;23:14133.
153. Osolodkin DI, Palyulin VA, Zefirov NS. Glycogen synthase kinase 3 as an anticancer drug target: novel experimental findings and trends in the design of inhibitors. Curr Pharm Des 2013;19:665-79.
154. Sahin I, Eturi A, De Souza A, et al. Glycogen synthase kinase-3 beta inhibitors as novel cancer treatments and modulators of antitumor immune responses. Cancer Biol Ther 2019;20:1047-56.
155. Augello G, Emma MR, Cusimano A, et al. The role of GSK-3 in cancer immunotherapy: GSK-3 inhibitors as a new frontier in cancer treatment. Cells 2020;9:1427.
156. Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen synthase kinase-3β participates in nuclear factor κB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res 2005;65:2076-81.
157. Ougolkov AV, Fernandez-Zapico ME, Bilim VN, Smyrk TC, Chari ST, Billadeau DD. Aberrant nuclear accumulation of glycogen synthase kinase-3β in human pancreatic cancer: association with kinase activity and tumor dedifferentiation. Clin Cancer Res 2006;12:5074-81.
158. Mai W, Miyashita K, Shakoori A, et al. Detection of active fraction of glycogen synthase kinase 3β in cancer cells by nonradioisotopic in vitro kinase assay. Oncology 2007;71:297-305.
159. Wilson W III, Baldwin AS. Maintenance of constitutive IκB kinase activity by glycogen synthase kinase-3α/β in pancreatic cancer. Cancer Res 2008;68:8156-63.
160. Mai W, Kawakami K, Shakoori A, et al. Deregulated GSK3 sustains gastrointestinal cancer cells survival by modulating human telomerase reverse transcriptase and telomerase. Clin Cancer Res 2009;15:6810-9.
161. Mamaghani S, Patel S, Hedley DW. Glycogen synthase kinase-3 inhibition disrupts nuclear factor-kappaB activity in pancreatic cancer, but fails to sensitize to gemcitabine chemotherapy. BMC Cancer 2009;9:132.
162. Gaisina IN, Gallier F, Ougolkov AV, et al. From a natural product lead to the identification of potent and selective benzofuran-3-yl-(indol-3-yl)maleimides as glycogen synthase kinase 3β inhibitors that suppress proliferation and survival of pancreatic cancer cells. J Med Chem 2009;52:1853-63.
163. Guzmán EA, Johnson JD, Linley PA, Gunasekera SE, Wright AE. A novel activity from an old compound: Manzamine A reduces the metastatic potential of AsPC-1 pancreatic cancer cells and sensitizes them to TRAIL-induced apoptosis. Invest New Drugs 2011;29:777-85.
164. Zhang JS, Koenig A, Harrison A, et al. Mutant K-Ras increases GSK-3β gene expression via an ETS-p300 transcriptional complex in pancreatic cancer. Oncogene 2011;30:3705-15.
165. Shimasaki T, Ishigaki Y, Nakamura Y, et al. Glycogen synthase kinase 3β inhibition sensitizes pancreatic cancer cells to gemcitabine. J Gastroenterol 2012;47:321-33.
166. Marchand B, Tremblay I, Cagnol S, Boucher MJ. Inhibition of glycogen synthase kinase-3 activity triggers an apoptotic response in pancreatic cancer cells through JNK-dependent mechanisms. Carcinogenesis 2012;33:529-37.
167. Zhou W, Wang L, Gou SM, et al. ShRNA silencing glycogen synthase kinase-3 beta inhibits tumor growth and angiogenesis in pancreatic cancer. Cancer Lett 2012;316:178-86.
168. Mamaghani S, Simpson CD, Cao PM, et al. Glycogen synthase kinase-3 inhibition sensitizes pancreatic cancer cells to TRAIL-induced apoptosis. PLoS One 2012;7:e41102.
169. Kitano A, Shimasaki T, Chikano Y, et al. Aberrant glycogen synthase kinase 3β is involved in pancreatic cancer cell invasion and resistance to therapy. PLoS One 2013;8:e55289.
170. Zhang JS, Herreros-Villanueva M, Koenig A, et al. Differential activity of GSK-3 isoforms regulates NF-κB and TRAIL- or TNFα induced apoptosis in pancreatic cancer cells. Cell Death Dis 2014;5:e1142.
171. Ying X, Jing L, Ma S, et al. GSK3β mediates pancreatic cancer cell invasion in vitro via the CXCR4/MMP-2 pathway. Cancer Cell Int 2015;15:70.
172. Kunnimalaiyaan S, Gamblin TC, Kunnimalaiyaan M. Glycogen synthase kinase-3 inhibitor AR-A014418 suppresses pancreatic cancer cell growth via inhibition of GSK-3-mediated Notch1 expression. HPB 2015;17:770-6.
173. Ma S, Li Q, Pan F. CXCR4 promotes GSK3β expression in pancreatic cancer cells via the Akt pathway. Int J Clin Oncol 2015;20:525-30.
174. Baumgart S, Chen NM, Zhang JS, et al. GSK-3β governs inflammation-induced NFATc2 signaling hubs to promote pancreatic cancer progression. Mol Cancer Ther 2016;15:491-502.
175. Liu B, Yang H, Pilarsky C, Weber GF. The effect of GPRC5a on the proliferation, migration ability, chemotherapy resistance, and phosphorylation of GSK-3β in pancreatic cancer. Int J Mol Sci 2018;19:1870.
176. Edderkaoui M, Chheda C, Soufi B, et al. An inhibitor of GSK3B and HDACs kills pancreatic cancer cells and slows pancreatic tumor growth and metastasis in mice. Gastroenterology 2018;155:1985-98.e5.
177. Kazi A, Xiang S, Yang H, et al. GSK3 suppression upregulates β-catenin and c-Myc to abrogate KRas-dependent tumors. Nat Commun 2018;9:5154.
178. Ding L, Madamsetty VS, Kiers S, et al. Glycogen synthase kinase-3 inhibition sensitizes pancreatic cancer cells to chemotherapy by abrogating the TopBP1/ATR-mediated DNA damage response. Clin Cancer Res 2019;25:6452-62.
179. Uehara M, Domoto T, Takenaka S, et al. Glycogen synthase kinase-3β participates in acquired resistance to gemcitabine in pancreatic cancer. Cancer Sci 2020;111:4405-16.
180. Carbone D, Parrino B, Cascioferro S, et al. 1,2,4-Oxadiazole topsentin analogs with antiproliferative activity against pancreatic cancer cells, targeting GSK3β kinase. ChemMedChem 2021;16:537-54.
181. Abrams SL, Akula SM, Meher AK, et al. GSK-3β can regulate the sensitivity of MIA-PaCa-2 pancreatic and MCF-7 breast cancer cells to chemotherapeutic drugs, targeted therapeutics and nutraceuticals. Cells 2021;10:816.
182. Palanivel C, Chaudhary N, Seshacharyulu P, et al. The GSK3 kinase and LZTR1 protein regulate the stability of Ras family proteins and the proliferation of pancreatic cancer cells. Neoplasia 2022;25:28-40.
183. Chikano Y, Domoto T, Furuta T, et al. Glycogen synthase kinase 3β sustains invasion of glioblastoma via the focal adhesion kinase, Rac1, and c-Jun N-terminal kinase-mediated pathway. Mol Cancer Ther 2015;14:564-74.
184. Kitabayashi T, Dong Y, Furuta T, et al. Identification of GSK3β inhibitor kenpaullone as a temozolomide enhancer against glioblastoma. Sci Rep 2019;9:10049.
185. Pothula SP, Xu Z, Goldstein D, Pirola RC, Wilson JS, Apte MV. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett 2016;381:194-200.
186. von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J Hematol Oncol 2017;10:76.
187. Helms E, Onate MK, Sherman MH. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov 2020;10:648-56.
188. Cao F, Li J, Sun H, Liu S, Cui Y, Li F. HES 1 is essential for chemoresistance induced by stellate cells and is associated with poor prognosis in pancreatic cancer. Oncol Rep 2015;33:1883-9.
189. Liu Y, Li F, Gao F, et al. Periostin promotes the chemotherapy resistance to gemcitabine in pancreatic cancer. Tumour Biol 2016;37:15283-91.
190. Zhang H, Wu H, Guan J, et al. Paracrine SDF-1α signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget 2015;6:3085-97.
191. Duluc C, Moatassim-Billah S, Chalabi-Dchar M, et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol Med 2015;7:735-53.
192. Cao H, Chu Y, Lv X, et al. GSK3 inhibitor-BIO regulates proliferation of immortalized pancreatic mesenchymal stem cells (iPMSCs). PLoS One 2012;7:e31502.
193. Bertrand FE. The cross-talk of NOTCH and GSK-3 signaling in colon and other cancers. Biochim Biophys Acta Mol Cell Res 2020;1867:118738.
194. Evangelisti C, Chiarini F, Paganelli F, Marmiroli S, Martelli AM. Crosstalks of GSK3 signaling with the mTOR network and effects on targeted therapy of cancer. Biochim Biophys Acta Mol Cell Res 2020;1867:118635.
195. Beurel E, Jope RS. Differential regulation of STAT family members by glycogen synthase kinase-3. J Biol Chem 2008;283:21934-44.
196. Beurel E, Jope RS. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J Neuroinflammation 2009;6:9.
197. Yoon J, Ko YS, Cho SJ, et al. Signal transducers and activators of transcription 3-induced metastatic potential in gastric cancer cells is enhanced by glycogen synthase kinase-3β. APMIS 2015;123:373-82.
198. Gao S, Li S, Duan X, et al. Inhibition of glycogen synthase kinase 3 beta (GSK3β) suppresses the progression of esophageal squamous cell carcinoma by modifying STAT3 activity. Mol Carcinog 2017;56:2301-16.
199. Sahin IH, Askan G, Hu ZI, O’Reilly EM. Immunotherapy in pancreatic ductal adenocarcinoma: an emerging entity? Ann Oncol 2017;28:2950-61.
200. Heumann T, Azad N. Correction to: Next-generation immunotherapy for pancreatic ductal adenocarcinoma: navigating pathways of immune resistance. Cancer Metastasis Rev 2021;40:863-4.
201. Hester R, Mazur PK, McAllister F. Immunotherapy in pancreatic adenocarcinoma: beyond “copy/paste”. Clin Cancer Res 2021;27:6287-97.
202. Bockorny B, Grossman JE, Hidalgo M. Facts and hopes in immunotherapy of pancreatic cancer. Clin Cancer Res 2022;28:4606-17.
203. Canel M, Sławińska AD, Lonergan DW, et al. FAK suppresses antigen processing and presentation to promote immune evasion in pancreatic cancer. Gut 2023;73:131-55.
204. Blanco-Gomez A, Jorgensen C. FAK scaffolds immune escape in pancreatic cancer. Gut 2023;73:6-8.
205. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol 2015;15:669-82.
206. Barrett RL, Puré E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. Elife 2020;9:e57243.
Cite This Article
 , ... Toshinari Minamoto
, ... Toshinari MinamotoHow to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright















Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].