


Hepatocellular carcinoma in alcoholic liver disease: mechanistic considerations and clinical facts


Abstract
Alcoholic hepatocellular carcinoma (AHCC) represents a lethal stage, emerging in the course of severe injurious stages of alcoholic liver disease including cirrhosis. AHCC only affects a few alcohol consumers, certainly not all individuals who consume large amounts of alcohol over a long period of time, suggesting a role of yet unknown genetic risk or protection factors. Most likely, hepatic DNA is ultimately involved, attacked by intermediate products derived from reactive oxygen species (ROS) generated from cytochrome P450 2E1 of the NADPH and oxygen dependent microsomal ethanol-oxidizing system whereby ethanol is metabolized. Ethanol and acetaldehyde are activated to procarcinogens, to be promoted to ultimate carcinogens by ROS and causatives for AHCC instead of any other putative chemical contained in alcoholic beverages. Prevention of HCC associated with cirrhosis is best accomplished by early recognition of alcohol abuse at the stage of alcoholic fatty liver rather than alcoholic hepatitis (AH) or alcoholic steatohepatitis (ASH), leading to the advice of consequent abstinence from alcohol. Abstinence early started effectively prevents AHCC development, as opposed to late begin of abstinence that lacks risk reduction. Although drug therapy may partially be effective in AH or ASH, no established drug options are available for a realistic therapy of AHCC. Liver transplantation is controversially discussed and can be considered, but may be an option for only a few patients on a case by case base. In conclusion, AHCC results from a ROS dependent conversion of ethanol and acetaldehyde to procarcinogens as promoters of AHCC.
Keywords
Introduction
The WHO considers cancer as the second leading cause of death globally, responsible for an estimated 9.6 million deaths in 2018, attributing about 1 in 6 deaths to cancer[1]. More specifically, the most common causes of cancer death are cancers of lung (1.76 million deaths), colon including rectum (862,000 deaths), stomach (783,000 deaths), liver (782,000 deaths), and breast (627,000 deaths), whereby around one third of deaths from cancer can be traced back to the 5 leading behavioral and dietary risks: high body mass index, low fruit and vegetable intake, lack of physical activity, tobacco use, and alcohol use[1]. Consequently, identifying the cause of liver cancer or more specifically primary hepatocellular carcinoma (HCC) in a patient with this type of cancer can be challenging and is variable among different countries or regions[2-7].
This article discusses current aspects of primary alcoholic hepatocellular carcinoma (AHCC) with focus on potential mechanistic steps and risk factors closely related to alcohol metabolism, reactive oxygen species (ROS), and the gut-liver axis in the context of the intestinal microbiome. In addition, clinical aspects with diagnostic approaches and therapy options including the controversially disputed liver transplantation will be considered.
Literature search and source
The PubMed database was used to identify publications for the following terms: alcohol, ethanol, DNA, ROS, alcoholic liver injury, and alcoholic hepatocellular carcinoma. Terms were used alone or combined. Limited to the English language, publications of the first 50 hits from each searched segment were analyzed for suitability of this review article. Publications were also derived from the large private archive. The search for publications was completed on 4 October 2019. The final compilation consisted of original papers, consensus reports, and review articles. The most relevant publications were included in the reference list of this review.
Alcohol as chemical
Ethanol is a short length chemical and a synonym to the term alcohol that is commonly used in a clinical setting. In the human gastrointestinal tract, alcohol can be produced from sugar with the help of intestinal bacteria and commonly undergoes rapid degradation by ubiquitous enzymes, for which alcohol serves as natural endogenous substrate keeping the respective enzymes active at low levels and prepared for larger amounts of exogenous alcohol eventually consumed as alcoholic beverage. Endogenous ethanol may be of clinical interest as “auto-brewery syndrome”, a drunk-driving defence challenge[8-10], with details provided in a case control study[10].
Clearly, alcohol is otherwise a product found in beverages such as wine prepared from the sugar containing grapes with the help of baker’s yeast alcohol dehydrogenase (ADH)[11,12]. Interestingly, a different ADH type is also present in humans and partly involved when the alcohol consumed as beverage is oxidized in the human gastrointestinal tract[13] and the liver[8,14,15].
Alcohol and acetaldehyde oxidation in the liver
After drinking of alcoholic beverages, alcohol is quickly taken up by the mucosa of the upper gastrointestinal tract[16], as evidenced by the prompt appearance in the blood with higher blood alcohol levels if the alcoholic beverage was consumed before a meal as long as the stomach is empty, as compared to lower blood alcohol levels found if the alcohol is consumed during or right after a meal[17]. As an exogenous compound lacking an option of storage within any organ, alcohol must be removed for which several possibilities exist[16]. Whereas only small amounts of the ingested alcohol leave the body by exhalation or with the urine, most of the alcohol must undergo enzymatic degradation in the liver[4,5,8,14-18]. The human liver is prepared to oxidize exogenous and endogenous alcohol using two different enzymes: ADH[5,8,13-17] and microsomal ethanol-oxidizing system (MEOS)[8,14-16,18-21]. Through both reactions, acetaldehyde is produced and its oxidation proceeds via the mitochondrial ALDH[8,15,16,18], requiring NAD+ as cofactor but it can also be metabolized in the endoplasmic reticulum of the liver[15].
ADH
Hepatic ADH is present in the cytosol of human liver cells and metabolizes ethanol according to the following equation:
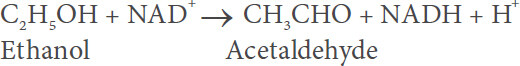
Additional information of ADH and its role in alcohol metabolism is provided in various other reports[5,8,13-17]. Evidence is lacking that in the course of this reaction reactive radicals are generated.
MEOS
MEOS is a constituent of the endoplasmic reticulum membranes in the human liver that correspond to the microsomal fraction following ultracentrifugation of the liver homogenate[8,14-16,18]. The oxidation of ethanol precedes via the following equation:
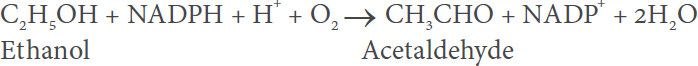
MEOS is different from ADH as well as catalase[19,20] and was isolated from these two enzymes using column chromatography[21-24], providing a typical elution pattern [Figure 1][21].

Figure 1. Purification of the MEOS and its separation from catalase and ADH activities. Separation was achieved by DEAE cellulose ion exchange column chromatography after solubilization of liver microsomes. In the void volume eluted up to around 220 mL, the highest peak represents the protein curve assessed as E280 nm, and the peak below that is the catalase peak, whereas ADH presents as the lowest peak. Starting with an elution volume of around 330 mL, microsomal components begin to appear. The first peak represents cytochrome P450, the second peak represents E280 nm, followed by a third peak with two shoulders and by a fourth peak representing MEOS. At around 770 mL, the reductase peak emerges, followed by the phospholipid peak at around 790 mL elution volume. Adapted from the original figure published in a previous report[21]. MEOS: microsomal ethanol-oxidizing system; ADH: alcohol dehydrogenase; DEAE: diethyl-amino-ethyl
Mechanistic consequences of alcohol metabolism related to carcinogenicity
Alcohol has a direct contact to the mucosal cells of the gastrointestinal tract with its MEOS and cytochrome P450 2E1 (CYP 2E1), and generated ROS may increase leakage of endotoxins, chemically known as lipopolysaccharides (LPS), out of the intestinal tract directed to the liver[8,15,16,25], modifying also the intestinal microbiome and the gut-liver axis. There is experimental and clinical evidence that Toll-like receptor 4 (TLR4) could be involved in AHCC development via signaling activation of LPS-TLR4[5,7], whereby coexistence of hepatitis B virus (HBV) an hepatitis C virus (HCV) infection could represent a risk factor[7].
Overall, alcohol dependent inflammation of the liver is responsible for non-maligant stages of alcoholic liver disease (ALD) but is a contributory factor of AHCC initiation through hepatocellular DNA damage[5]. Specific liver macrophages are involved in AHCC development triggered by activated hepatic Stellate cells (HSCs)[5], which are also known as promoters of liver alcoholic fibrosis[8,15]. Ectopic expression of TLR4 in hepatocytes and its activation by LPS triggers AHCC through production of TLR4 and tumor-initiating stem-cell like cells[5]. It has been reported that activated HSCs may also promote AHCC development via matrix or soluble factors that help tumor cell survival and growth[5].
Most importantly, ROS can bind to and damage DNA and cause lipid peroxidation with the generation of highly carcinogenic exocyclic etheno-DNA adducts[5]. Other DNA adducts of interest include N2-ethyl-deoxyguanosine and N2-propano-2’-deoxyguanosine, which may modify the DNA integrity, whereas acetaldehyde protein adducts may impair the DNA repair system[7]. Consequently, the direct DNA mutagenic effect of acetaldehyde and the indirect carcinogenic effect of adducts may modulate carcinogenesis leading to AHCC[5,6,26,27]. Other considerations focus on DNA methylation that modifies the expression of genes: hypomethylation is related to enhanced gene expression, considered as an import factor of cancer development[6]. Prolonged alcohol use also causes a reduction of S-adenosylmethionine in the liver, the methyl donor of DNA[6]. Reviewing the relevant literature with inclusion of those referenced[5-7], it is obvious that many mechanism have the potential of causing AHCC. In face of multiple proposals and study data, and as expected, not all results are confirmative but even contradictory, not allowing a valid uniform proposal how AHCC develops in the human liver during prolonged alcohol abuse.
Principles of hepatic carcinogenesis
There is general believe that carcinogenesis of various organs including the liver is triggered by an enhancement of oncogene expression or due to an impairment of cells to improve their DNA quality leading to inappropriate DNA repair associated with DNA mutations, conditions that facilitate oncogenic mutations[5,6]. Conditions in the human and experimental setting of AHCC are complex due to a broad range of variabilities. Although these variable DNA related modifications had been observed mostly in animal models or human tissues, respective translation of these meaningful data to humans with AHCC is warranted. The variability of proposed molecules damaging DNA as a result of alcohol consumption and metabolism requires a closer look with focus on alcohol metabolism and more specifically electrophile metabolites, in addition to the concomitant production of ROS to be discussed in general.
Microsomal components of MEOS
ROS production is closely connected with the degradation of ethanol via MEOS, which is not a single enzyme but represents a system composed of the three microsomal components CYP 2E1, the reductase, and phospholipids without their yet clearly determined place of interaction [Figure 2], as early recognized during isolation of MEOS [Figure 1][21], with ROS production and ethanol oxidation carried out by the three components[8,15,16,18]. Separated from ADH and catalase activities shown on the left side of the elution pattern, MEOS activity was recovered in eluates on the right side containing all three microsomal constituents: cytochrome P450, NADPH-cytochrome c reductase (better to be analyzed as compared to NADPH cytochrome P450 reductase), and microsomal phospholipids[21].

Figure 2. Constituents of the microsomal ethanol-oxidizing system
Using our published elution procedure of a stepwise KCl gradient[22], a subsequent report reaffirmed the validity and reproducibility of our initial results in all details[28], as published before[22]. This reassured our proposed participation of special microsomal enzymes and constituents in MEOS and its independence from ADH and catalase. The participation and obligatory role of these three components for MEOS was later verified using purified microsomal constituents in order to reconstitute MEOS[29,30], in support by other studies showing the direct oxidation of ethanol by a catalase-free and alcohol dehydrogenase-free reconstituted system containing cytochrome P-450[31]. Therefore, a close association between ROS and MEOS exists, to be further discussed in relation to carcinogenicity and AHCC.
Reactive oxygen species
New sophisticated analytical techniques were developed that helped characterize MEOS, CYP isoforms with preference of microsomal CYP 2E1, and ROS[4,8,15,16,18,32-36]. One of the highlights was the observation in humans that MEOS consists of several CYP isoforms with CYP 2E1 as the most active in microsomal ethanol oxidation[32], with variable turnover numbers of MEOS activity if calculated per CYP isoform content [Table 1][32].
MEOS and its cytochrome P450 isoforms
| Cytochrome P450 isoforms | MEOS activity/cytochrome P450 |
|---|---|
| 1A2 | 10.90 |
| 2A6 | 3.75 |
| 2B6 | 2.89 |
| 2D6 | 0.70 |
| 2E1 | 11.51 |
| A4 | 3.38 |
With focus on the hepatic microsomal CYP 2E1, a variety of phenomena can now be explained, as illustrated with a few examples[8,15,16,19-21,28,32-39]: (1) prolonged alcohol consumption upregulates the NADPH and oxygen dependent MEOS activity[19-21,37,38] that adaptively enhances the overall metabolism of ethanol[36,37] through an increase of CYP 2E1[29,33]; (2) in the presence of NADPH and molecular oxygen, CYP 2E1 metabolizes not only ethanol in the context of MEOS but also many other exogenous substrates with metabolic competitive interactions at the level of CYP [Figure 3][16]; (3) during CYP 2E1 dependent reactions, oxygen split often is incomplete [Figure 4][16], allowing the generation of various types of ROS and ROS dependent products [Table 2][16]; (4) part of the ROS will be used to metabolize ethanol or acetaldehyde to reactive intermediates, whereby initially ethanol and acetaldehyde per se are not carcinogens but can be classified as procarcinogens that are converted to ultimate carcinogens with the potential of inducing AHCC by attacking DNA[39]; and (5) in addition, parts of ROS not used for MEOS would be freely available for direct attack of DNA and could also trigger radical formation of soluble proteins and or phospholipids or those located in structural membranes of liver cells organelles including mitochondria[8,15,16,18,33-36,39].

Figure 3. Microsomal metabolism of various chemicals including ethanol. At the level of cytochrome P450, metabolic drug-drug and drug-ethanol interactions are feasible. Figure derived from a previous report[15]

Figure 4. Hepatic microsomal cytochrome P450 and its interaction with substrates. Cytochrome P450 catalyzes the oxidation of substrates such as drugs and ethanol, which bind to the ferric (3+) iron of the cytochrome P450 as the initial metabolic step leading finally to the oxidized substrate. The original figure was published in and derived from a recent article[15]
Potentially toxic metabolites resulting from enzymatic degradation of ethanol in the liver
| Selected potentially toxic metabolites and reactive O2-species due to hepatic ethanol degradation |
|---|
| Acetaldehyde C2H4O |
| Ethoxy radical CH3CH2O• |
| Hydroxyethyl radical CH3C(•)HOH |
| Acetyl radical CH3CHO• |
| Singlet radical 1O2 |
| Superoxide radical HO•2 |
| Hydrogen peroxide H2O2 |
| Hydroxyl radical HO• |
| Alkoxyl radical RO• |
| Peroxyl radical ROO• |
| Lipidperoxides |
Putative procarcinogens and ultimate carcinogens
As a reminder, CYP 2E1 metabolizes not only alcohols but also a bundle of other substrates, some of these are carcinogens or procarcinogens [Table 3], with references for each listed chemical provided in a previous publication[16]. Abundant clinical and experimental publications relate to alcohol and liver cancer but further specification is often missing and major questions remain: (1) are alcoholic beverages globally carcinogenic or only specific ingredients like contaminating carcinogens? or (2) is the chemical ethanol per se carcinogenic or is it merely a procarcinogen? or (3) is acetaldehyde as the first metabolite in its original form and per se carcinogenic or is metabolic activation to a reactive intermediate prerequisite to be classified as procarcinogen? or (4) can ROS be considered as carcinogen either alone or perhaps together with an activated membrane protein or a membrane phospholipid that can easily be converted to a reactive lipidperoxide, considering that ROS alone in the absence of alcohol may play a role for HCC in nonalcoholic steatohepatitis (NASH).
Selected substrates of the hepatic microsomal CYP 2E1
| Selected substrates catalyzed by CYP 2E1 |
|---|
| Acetaldehyde |
| Acetol |
| Acetone |
| Acetaminophen |
| Aniline |
| Benzene |
| Bromobenzene |
| n-Butanol |
| Caffeine |
| Carbon tetrachloride |
| Chloroform |
| 1-Chloropropane |
| Chlorzoxazone |
| 1,1-Dichloroethane |
| 1,2-Dichloroethane |
| 1,1-Dichloroethylene |
| cis-1,2-Dichloroethylene |
| trans-1,2-Dichloroethylene |
| Dichloromethane |
| 1,2-Dichloropropane |
| 1,2-Dibromoethane |
| Diethylether |
| Dimethylformamide |
| Cumene |
| Enflurane |
| Ethanol |
| Ethylbenzene |
| Halothane |
| n-Hexane |
| Isoflurane |
| Methanol |
| Methyl t-butyl ether |
| Methoxyflurane |
| Monochlorobenzene |
| 4-Nitrophenol |
| p-Nitrophenol |
| Nitrosamines |
| N-nitrosodimethylamine |
| n-Pentane |
| Phenol |
| n-Propanol |
| Propylbenzene |
| Sevoflurane |
| Styrene |
| 1,1,1,2-Tetrachloroethane |
| 1,1,2,2-Tetrachloroethane |
| Tetrachloroethylene |
| Toluol |
| Trichloroethylene |
| 1,1,1-Trichloroethane |
| 1,1,2-Trichloroethane |
| Vinylchloride |
| o,m,p-Xylene |
Officially, alcohol is now classified as a human carcinogen rather than as a clear procarcinogen by the International Agency for Research in Cancer in a global context[5,39-41], disregarding the known complexity of metabolic events leading to a variability of compounds as outlined above.
Cocarcinogens
Humans with an alcohol problem are often heavy smokers[42] and confronted with potential mutagenic and carcinogenic compounds in the tobacco smoke may enter the body and act as cocarcinogens[43]. On theoretical backgrounds but difficult to evaluate in patients, mutagenic and carcinogenic compounds may contaminate alcoholic beverages and could function upon ingestion as cocarcinogens, contributing to initiation and promotion of AHCC[41]. However, in the majority of alcoholic beverages mutagenic and carcinogenic chemicals are not found, but if detected, respective amounts are very small and unlikely causing harm the liver. In experimental studies, tumor incidence in the liver caused by dimethylnitrosamine (DMN) is influenced by prolonged alcohol use as evidenced by a specific study protocol[44]: rats were pair-fed for 3 weeks a nutritionally adequate liquid diet containing either ethanol (36% of total calories) or isocalorically substituted carbohydrates as control diet. Thereafter, the animals were maintained on laboratory chow and tap water ad libitum for another 2 weeks and received 1.5 mg DMN intraperitoneally per day for the first 5 days. This 5-week cycle was repeated three more times. Chronic pretreatment with the alcohol-containing diet significantly improved the mean survival time of DMN-treated rats compared with identically treated animals fed before with the control diet, but the total number of tumors observed under these experimental conditions and the target organ remained virtually unchanged[44]. The partially positive and protective effect on survival in DMN treated animals elicited by prolonged alcohol consumption was unexpected, as was the reduced liver injury by DMN following prolonged pretreatment with the alcohol containing diet[45,46].
Cascades of events: from liver injury and alcoholic fatty liver to AHCC
The spectrum of ALD is broad and well described in various reports provided by experts in the field[2,5-8,15-17,47-59], starting with the most frequent stage of alcoholic fatty liver (AFL)[47-49], and the transition to alcoholic steatohepatitis (ASH) and alcoholic hepatitis (AH), both of which follow different criteria[50-53], with alcoholic cirrhosis (AC), the most known end stage of ALD[54-57] and often precursor of AHCC[2,5-8,58,59]. Delineating a 5-hit proposal has the advantage of a clear structure and better overview and has been published previously [Figure 5][8].

Figure 5. The 5-hit working hypothesis in alcoholic liver disease. The 5-hit hypothesis presents various possible steps leading from alcoholic fatty liver, eventually to hepatocellular carcinoma. In clinical practice, some patients with alcoholic hepatitis do not have steatosis/steatohepatitis as a precursor, with additional details provided in Table 4. The original figure was published in an earlier report[15]
Alcoholic liver disease and the 5-hit working hypothesis with a tentative cascade of events
| First hit | The first hit is dependent on ADH and occurs at low alcohol levels through the generation not only of NADH + H+ leading to an increased NADH + H+/NAD+ ratio, which stimulates hepatic fatty acid synthesis[22] and increases α-glycerophosphate-trapping fatty acids, but also of acetaldehyde, which impairs hepatic mitochondrial functions including hepatic mitochondrial fatty acid oxidation. This first hit fully explains at least in part the development of alcoholic fatty liver. |
| Second hit | The second hit is classified as a transition from alcoholic fatty liver to alcoholic steatohepatitis, most likely triggered by the increased production of acetaldehyde via MEOS, and of ROS with its capacity for irreversible covalently binding to cellular macromolecules, including membrane proteins and phospholipids. These injurious alterations at the molecular and cellular level cause some necrosis, apoptosis, and inflammatory cells in the fatty liver, justifying the term alcoholic steatohepatitis, as it includes toxic hepatitis in steatosis. Further stages are characterized by perisinusoidal and pericentral fibrosis due to participation of non-hepatocytes such as Kupffer cells, stellate cells, and sinus endothelial cells. Mediators such growth factors, interferons, interleukins, tumor necrosis factor and endotoxins, as well as hepatic iron, are considered as possible active promoters of liver injury, but considering the multiplicity of proposed mediators, it is difficult to predict how they interact with each other and modify the course of liver injury. |
| Third hit | The third hit initiates a more severe liver injury stage, whereby alcoholic steatohepatitis is the precursor in most, but certainly not all patients with alcoholic hepatitis. Steatosis is no more a characteristic feature, but is now replaced by necrosis, apoptosis, and inflammation. At this stage, injury becomes more severe and presents with more fibrosis and as a self-perpetuating process, immunity aspects gain additional relevance, because alcohol modifies the innate and adapted immune system, which may explain the individual differences of susceptibility for ALD. With the third hit, the disease may approach a point of no return. |
| Fourth hit | The fourth hit is dominated by increased fibrosis, due to increased collagen formation. This allows for a clinically unrecognizable transition from alcoholic hepatitis with fibrosis to irreversible cirrhosis. However, AC can also develop without ASH or AH. |
| Fifth hit | In rare cases, a fifth hit initiates the development of a HCC, mostly occurring in patients with cirrhosis. This final hit scenario of carcinogenesis is triggered by acetaldehyde and ROS through the generation of DNA adducts, which promote mutagenesis, and interference with methylation, synthesis, and repair of DNA. Suggested is a possible role of SIRT1. These overall events will enhance AHCC susceptibility, especially in the absence of an identifiable carcinogen. |
All 5-hit stages are well defined by both, clinical and histology evaluations, but some overlap among the stages is unavoidable. Pathogenic details of all ALD stages have been published in a recent review article[8], some additional details are presented for AFL, ASH, AH, AC and AHCC for a brief overview [Table 4].
Clinical challenges of AHHC
Characteristic clinical features
AHCC develops mostly in patients with existing AC that may be compensated or not, and the degree of decompensation of AC will determine clinical symptoms superimposing those of AHCC. Clinical symptoms of patients with AHCC have not systematically been investigated and published recently[5-8,15,58,59], but based on own clinical experience, patients with AHCC may have a silent clinical course until decompensation of AC develops. Until that point, patients are either without symptoms or report unspecific signs like fatigue, weakness, loss of appetite and weight, nausea and vomiting, and upper abdominal pain.
Pathology
Histology
The histologic findings of all stages in ALD have comprehensively been outlined[2], with some specificities regarding HCC and its variants[60-62]: the steatohepatic HCC variant is characterized by a steatotic appearance of > 5% of the tumor, presence of Mallory bodies, fibrosis, inflammation and ballooning of the hepatocytes as in steatohepatitis[60]. The inflammatory infiltrate usually consists of neutrophils, plasma cells, and lymphocytes. Microscopically, cells of classical HCC resemble normal liver cells, the similarity to normal liver is most evident in well to moderately differentiated tumors, but liver cell plates change from 1 to 2 cell nuclei to 3 or more[62]. In particular, AHCCs with a diameter of 1.5 to 2 cm are usually moderately or well differentiated with a distinct nodular structure, a rate of microinvasion of between 10% and 22%, with satellitosis in around 10%[6].
Gross pathology
Macroscopically, AHCC is commonly seen in a cirrhotic liver but around 10% of AHCC are found in alcoholic patients without AC or liver fibrosis[59]. AHCC nodules may present singular, or multiple as illustrated by the liver of a patient with a long history of alcohol consumption [Figure 6]. The multiple larger nodes of AHCC stretched multifocal over the liver that otherwise presents as typical AC with focal, small regenerative cirrhotic nodules, mostly of the micronodular type, better seen in another picture presenting only AC that shows light splitting in the areas of micronodular regenerating nodules [Figure 7][15].

Figure 6. Macroscopic picture of alcoholic hepatocellular carcinoma

Figure 7. Macroscopic picture of alcoholic cirrhosis
Risk factors
Smoking
Sufficient evidence supports the view that smoking strikingly increases the risk of AHCC[43], an important issue stressed also in a recent review article[5].
Acquired comorbidities
Risk factors for AHCC include not only AC but also cirrhosis of various other causes and chronic co-infection by hepatitis B or C[5,59]. Indeed, a broad range of confounding variables exists that can contribute to the development of AHCC[5,43,59]. Among these are diabetes mellitus with the metabolic syndrome, NASH or nonalcoholic fatty liver disease due to overweight or morbid obesity[5,59]. In this context, not only ROS generated in the fat tissues and the liver[5,59,63-65] but also lipotoxicity in general and more specifically as lipotoxic liver injury[65] may represent additional risk factors for causing AC and AHCC[5].
Genetic diseases
Genetic liver diseases like α1-antitrypsin (AT) deficiency leading to storage of AT in the liver, Wilson disease with Cu storage in the liver, or the primary hereditary hemochromatosis (HH) with Fe storage in the liver, all these inherited diseases may lead per se to liver cirrhosis, and HCC are specific risk factors. They are risky due to their existence starting at birth and preexisting mostly for many years before alcohol abuse is initiated. HH is of special interest due to its high Fe content in the liver. From a metabolic aspect, Fe was early recognized in unpublished studies as promoter of MEOS activity, which led to the addition of EDTA to each MEOS assay to capture any Fe in the assay system[22-24]. Subsequent studies have confirmed that Fe stimulates not only MEOS activity but also ROS including hydroxyl radical generation[36]. This mechanism could explain why the risk of AHCC is increased in patients with HH who abuse alcohol for a long time[66], substantiated by a subsequent analysis of available data, which suggest that iron accumulation in the liver is an independent risk factor for HCC in patients with AC[67]. In particular, iron accumulation in the liver is considered to be a co-factor for progression of liver disease, and iron overload can enhance the effects of oxidative stress and influence the natural history of patients with cirrhosis, exposing them to a higher risk of HCC.
Gene polymorphisms
Enzymes metabolizing ethanol and acetaldehyde in the liver are individually characterized and modified by gene polymorphisms[7,8,15], with abundant studies that addressed the relevance of respective gene polymorphisms for their risk of AHCC[7]. However, results were contradictory. With ADH and ALDH studied in a Japanese cohort as an example, gene polymorphisms of ADH2 and ALDH2 were found to correlate with AHCC development[7], findings not confirmed subsequently[7,68].
Gender
Overall prevalence of AHCC is small in women compared with that in men[7]. Considering this limitation and a subgroup of consumers who used more than 80 g alcohol per day, the risk of AHCC development was fivefold higher in women than in men[7,69]. In general, female alcoholic patients are at a higher risk for ALD as compared to alcoholic men[5,8,70,71]. In particular, women have more advanced liver disease at time of diagnosis, experience a more severe clinical course within a shorter time of alcohol abuse, and had consumed less alcohol compared to men[72], in line with a lower thresholds for development of alcoholic liver injury[71,72]. This gender difference can be traced back to higher blood alcohol concentrations in woman compared to men who consume the same amount of alcohol, resulting from a lower proportion of body water in females than in males of equal body weight[50] and from a lower ADH dependent first pass metabolism in the gastric mucosa[73]. Under discussion are also gender based differences in the sensitivity of hepatic Kupffer cells to endotoxins generated in the gut[50].
Immune system
Little is firmly established related to specific immune reactions that could assist initiate and perpetuate AHCC although immune involvement is most likely[7], as known from other tumors. Alcohol can modify both, the innate immune system (IIS) and the adaptive immune system (AIS)[8,15]. More specifically, IIS is promoted by macrophages, Kupffer cells, neutrophils, and natural killer cells, whereby macrophages are prepared to attack antigens of bacterial cell walls and respond by providing cytokines.
Amount of alcohol
An excessive alcohol consumption is a risk factor not only of AC[7,15] but also for AHCC[7]. For both stages of ALD, a rough linear dose-response relationship between the amount of alcohol consumption and the respective risk is assumed[7]. For AHCC, an alcohol use expressed as absolute ethanol is risky at and above > 60-100 g per day, that compares with an odd ratio of 4.52 if the total amount of alcohol consumption during lifetime is considered.
Diagnosis of AHCC and surveillance programmes
As an inexpensive method readily available in clinical settings, ultrasound is commonly used to diagnose AC and AHCC[74,75]. Ultrasound parameters for AC include liver size, bluntness of the edge, coarsened of the liver parenchyma, nodularity of the liver surface, and spleen size[74], while data of liver stiffness could be supportive[5]. In search for AHCC, ultrasound technique has been improved by introducing the contrast-enhanced ultrasound (CEUS), considered as a major diagnostic breakthrough[75]. CEUS is unique in that it allows non-invasive assessment of liver perfusion in real time throughout the vascular phase and may abandon previous methods such as magnetic resonance or computer tomography images. Under imaging guidance, tissue from the suspected HCC may be obtained to verify histologically the diagnosis.
In line with other causes of cirrhosis, periodic screening for AHCC is recommended for patients with AC, who should be included in respective surveillance programmes[59], which offer screenings at intervals of 6 months. This allows early detection of AHCC and implementation of curative procedures. However, contradictory recommendations suggest surveillance by biannual ultrasound[76].
Prevention
Clinical medicine should focus primarily on prevention of AHCC for the sake of the patient at risk, the human society, and financials of the health system. Overall prevention of AHCC is most successful if alcohol abuse can early be stopped. First of all, if physicians suspect that a patient may have an alcohol problem, specific questionnaires could help rule out or confirm the problem, as summarized recently[53]. A better and more objective approach would be doing just two laboratory tests in search for a serum quotient AST/ALT that may be > 1.0 if an alcohol problem exists independently from a specific ALD stage including already AFL[15]. The ratio is significantly increased in patients with AH and AC (2.85 ± 0.2). This led to the proposal that a serum AST/ALT ratio > 2.0 is highly suggestive of alcoholic hepatitis and cirrhosis[15,77]. Values of the AST/ALT ratio have been published for patients with AFL (1.64 ± 1.57), as compared with a corresponding control group consisting of individuals with normal liver histology and normal values of AST and ALT, showing a lower AST/ALT ratio (0.72 ± 0.24)[15,78]. In search for individuals with severe alcohol abuse, other laboratory data show variable percentages of sensitivity: carbohydrate-deficient transferrin (CDT; 63%), gamma-glutamyltransferase (GGT; 58%), mean corpuscular volume of erythrocytes (MCV; 45%), aspartate aminotransferase (AST; 47%), alanine aminotransferase (ALT; 50%), and GGT + CDT (90%)[15].
Under the aspect of prevention and associated diagnosis, a liver biopsy to establish the diagnosis of any ALD stage is certainly not recommended under routine conditions, although it was previously considered as diagnostic gold standard. Exemptions now are pretransplantation evaluations or RCTs to test efficacy of drugs for instance in patients with AH, but in most other cases the patient will have no benefit from this invasive procedure. This is also confronted with a fatality rate, though rather low[5].
Pharmacotherapy option
If AHCC is diagnosed, the patient should abstain from further alcohol use to prevent destruction of the remaining, still functioning liver. While some pharmacotherapy measures are partially effective in patients with severe AH[53], respective options with proven efficacy are not available for AHCC. Instead, palliative measures for pain and symptom relief is the only choice.
Segment resection, liver transplantation, and tumor ablation
A stringent algorithm for management of AHCC has been presented that should be used as a guideline[66]. Options must consider specific criteria and may include segment resection, disputed liver transplantation, locoregional ablation using Sorafenib, transarterial chemoembolization, and radiotherapy. Investigational studies should be performed in the frame of RCTs.
Prognosis
Presenting mostly as an end stage of the tumor disease, patients with AHCC have a poor prognosis, because overall survival was in a range of 15 to 32 months, which compared to 16 to 47 months in patients who received a curative treatment[59]. This again calls for an early recognition of potential individuals with an alcohol problem and associated early stages of ALD. Vague estimates based on four studies[79] allow the tentative conclusion that abstinence from alcohol use may reduce AHCC development perhaps by 6% to 7% per year, but a wash out period of 23 years being necessary to achieve the same incidence of AHCC seen in abstinent individuals[59,79]. In other words, early abstinence is better than late abstinence.
Epidemiology AHCC
Worldwide epidemiology data of AHCC suggests that alcohol accounts for around one third of global incident cases of primary liver cancer, with a substantial variability of results among different countries and regions, ranging from 6% in Iran to 61% in Estonia and Moldova[59,80]. Details can be derived from a recent large study of the Global Burden of Disease Liver Cancer Collaboration, which considers the burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level[80]. Accordingly, for virtually all countries data of AHCC and liver cancer due to HBV, HCV, and other causes (OC) are available, for example[80]: Australia (AHCC 38%, HBV 9%, HCV 40%, OC 13%), Austria (AHCC 49%, HBV 15%, HCV 30%, OC 6%), Belgium (AHCC 48%, HBV 16%, HCV 28%, OC 8%), China (AHCC 33%, HBV 41%, HCV 8%, OC 18%), France (AHCC 37%, HBV 17%, HCV 36%, OC 9%), Germany (AHCC 44%, HBV 8%, HCV 33%, OC 14%), India (AHCC 21%, HBV 42%, HCV 20%, OC 18%), Japan (AHCC 17%, HBV 8%, HCV 69%, OC 6%), Russia (AHCC 53%, HBV 15%, HCV 24%, OC 8%), United Kingdom (AHCC 36%, HBV 17%, HCV 38%, OC 9%), and United States (AHCC 37%, HBV 9%, HCV 31%, OC 22%)[80]. In countries with a high AHCC percentage, a high alcohol consumption is likely. In other countries, HBV and HCV infections are causative for HCC.
Conclusions
Ethanol per se is not carcinogenic but has to be viewed as a procarcinogen involved in the development of AHCC. This process is triggered by ROS including activated molecules derived from ethanol or acetaldehyde metabolism attacking hepatocellular DNA. Upregulation of ROS production occurs via CYP 2E1 following prolonged alcohol consumption and represents a major risk factor of AHCC. To prevent AHCC, individuals with an alcohol problems must early be identified in order to achieve alcohol abuse, because only a long period of abstinence will substantially reduce the risk of AHCC initiation, short term abstinence contributes not or little to risk reduction. Concomitant chronic liver diseases are additional risks factors in the context of AHCC and deserve effective treatment. Of special risks are infections by HBV and HCV and genetic liver disease such as hereditary hemochromatosis with its high Fe content in the liver. AHCC represents a late stage of ALD mostly in connection with AC and does not provide early clinical warning symptoms.
Declarations
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. WHO. Cancer. Available from: https://www.who.int/news-room/fact-sheets/detail/cancer. [Last accessed on 6 Nov 2019].
3. Rawla P, Sunkara T, Muralidharan P, Raj JP. Update in global trends and aetiology of hepatocellular carcinoma. Contemp Oncol (Pozn) 2018;22:141-50.
4. Wu D, Cederbaum AI. Alcohol, oxidative stress, and free radical damage. Available from: https://pubs.niaaa.nih.gov/publications/arh27-4/277-284.htm. [Last accessed on 6 Nov 2019].
5. Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, et al. Alcoholic liver disease. Nat Rev Dis Prim 2018;4:16.
6. Testino G, Leone S, Borro P. Alcohol and hepatocellular carcinoma: a review and a point of view. World J Gastroenterol 2014;20:15943-54.
8. Teschke R. Alcoholic liver disease: current mechanistic aspects with focus on their clinical relevance. Biomedicines 2019;7:68.
9. Logan BK. Endogenous ethanol “auto-brewery syndrome” as a drunk-driving defence challenge. Med Sci Law 2000;40:206-15.
10. Cordell BJ, Kanodia A, Miller GK. Case-control research study of auto-brewery syndrome. Global Adv Health Med 2019;8:1-7.
11. Jolly NP, Augustyn OPH, Pretorius IS. The role and use of the non-Saccharomyces yeasts in wine production. S Afr J Enol Vitic 2006;27:15-39.
12. Moreno-Arribas MV, Polo MC. Wine Chemistry and Biochemistry. Springer New York; 2009.
13. Seitz HK, Egerer G, Simanowski UA, Waldherr R, Eckey R, et al. Human gastric alcohol dehydrogenase activity: effect of age, gender and alcoholism. Gut 1993;34:1433-7.
15. Teschke R. Alcoholic liver disease: alcohol metabolism, cascade of molecular mechanisms, cellular targets, and clinical aspects. Biomedicines 2018;6:106.
16. Teschke R. Biochemical aspects of the hepatic microsomal ethanol-oxidizing system (MEOS): resolved initial controversies and updated molecular views. Biochem Pharmacol (Los Angel) 2019;8:267.
17. Salaspuro M. Epidemiological aspects of alcohol and alcoholic liver disease, ethanol metabolism, and pathogenesis of alcoholic liver injury. In: Bircher J, Benhamou JP, McIntyre N, Rizzetto N, Rodes J, editors. Oxford Textbook of Clinical Hepatology. Oxford University Press, Oxford; 1999. pp. 791-810.
18. Teschke R. Microsomal ethanol-oxidizing system (MEOS): success over 50 years and an encouraging future. Alcohol Clin Exp Res 2019;43:386-400.
19. Lieber CS, DeCarli LM. Ethanol oxidation by hepatic microsomes: adaptive increase after ethanol feeding. Science 1968;162:917-8.
20. Lieber CS, DeCarli LM. Hepatic microsomal ethanol-oxidizing system. In vitro characteristics and adaptive properties in vivo. J Biol Chem 1970;245:2505-12.
21. Teschke R, Hasumura Y, Joly JG, Ishii H, Lieber CS. Microsomal ethanol-oxidizing system (MEOS): purification and properties of a rat liver system free of catalase and alcohol dehydrogenase. Biochem Biophys Res Commun 1972;49:1187-93.
22. Teschke R, Hasumura Y, Lieber CS. Hepatic microsomal ethanol oxidizing system: solubilization, isolation and characterization. Arch Biochem Biophys 1974;163:404-15.
23. Teschke R, Hasumura Y, Lieber CS. NADPH-dependent oxidation of methanol, ethanol, propanol, and butanol by hepatic microsomes. Biochem Biophys Res Commun 1974;60:851-7.
24. Teschke R, Hasumura Y, Lieber CS. Hepatic microsomal alcohol-oxidizing system: affinity for methanol, ethanol, propanol and butanol. J Biol Chem 1975;250:7397-404.
25. Teschke R, Zhu Y. Opinion: intestinal microbiome, endotoxins, cytochrome P450 2E1, and the gut-liver axis in alcoholic liver disease EC Gastroenterology Dig Syst. 2019;6.2:66-75. Available from: https://www.ecronicon.com/ecgds/pdf/ECGDS-06-00312.pdf [Last accessed on 6 Nov 2019].
26. Hasumura Y, Teschke R, Lieber CS. Characteristics of acetaldehyde oxidation in rat liver mitochondria. J Biol Chem 1976;251:4908-13.
27. Hasumura Y, Teschke R, Lieber CS. Acetaldehyde oxidation by hepatic mitochondria: its decrease after chronic ethanol consumption. Science 1975;189:727-9.
28. Damgaard SE. The D(V/K) isotope effect of the cytochrome P-450-mediated oxidation of ethanol and its biological applications. Eur J Biochem 1982;125:593-603.
29. Ohnishi K, Lieber CS. Reconstitution of the microsomal ethanol-oxidizing system. Qualitative and quantitative changes of cytochrome P-450 after chronic ethanol consumption. J Biol Chem 1977;252:7124-31.
30. Teschke R, Ohnishi K, Hasumura Y, Lieber CS. Hepatic microsomal ethanol oxidizing system: isolation and reconstitution. In: Ullrich V, Roots I, Hildebrandt A, Estabrook RW, Conney AH, editors. Microsomes and Drug Oxidations. Pergamon Press, Oxford; 1977. pp. 103-10.
31. Miwa GT, Lewin W, Thomas PE, Lu AY. The direct oxidation of ethanol by a catalase-free and alcohol dehydrogenase-free reconstituted system containing cytochrome P-450. Arch Biochem Biophys 1978;187:464-75.
32. Asai H, Imaoka S, Kuroki T, Monna T, Funnae Y. Microsomal ethanol oxidizing system activity by human hepatic cytochrome P-450s. J Pharmacol Exp Ther 1996;277:1004-9.
33. Lieber CS. Cytochrome P-4502E1: its physiological and pathological role. Physiol Rev 1997;77:517-44.
34. Cederbaum AI, Dicker E, Rubin E, Lieber CS. The effect of dimethylsulfoxide and other radical scavengers on the oxidation of ethanol by rat liver microsomes. Biochem Biophys Res Commun 1977;78:1254-62.
35. Ohnishi K, Lieber CS. Respective role of superoxide and hydroxyl radical in the activity of the reconstituted microsomal ethanol-oxidizing system. Arch Biochem Biophys 1978;191:798-803.
36. Ingelman-Sundberg M, Johansson I. Mechanisms of hydroxyl radical formation and ethanol oxidation by ethanol-inducible and other forms of rabbit liver microsomal cytochrome P-450. J Biol Chem 1984;259:6447-58.
37. Lieber CS, DeCarli LM. The role of the hepatic microsomal ethanol-oxidizing system (MEOS) for ethanol metabolism in vivo. J Pharmacol Exp Ther 1972;181:279-87.
38. Lieber CS, DeCarli LM, Matsuzaki S, Ohnishi K, Teschke R. The microsomal ethanol-oxidizing system. In: Fleischer S, Packer L, editors. Methods in Enzymology. Academic Press: New York; 1978. pp. 355-68.
39. Brooks PJ. DNA damage, DNA repair, and alcohol toxicity - a review. Alcohol Clin Exp Res 1997;6:1073-82.
40. Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, et al; WHO International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of alcoholic beverages. Lancet Oncol 2007;8:292-3.
41. Lachenmeier DW, Przybylski M, Rehm J. Comparative risk assessment of carcinogens in alcoholic beverages using the margin of exposure approach. Int J Cancer 2012;131:E995-1003.
42. Hart CL, Davey Smith G, Gruer L, Watt GC. The combined effect of smoking tobacco and drinking alcohol on cause-specific mortality: a 30 year cohort study. BMC Public Health 2010;10:789.
43. Purohit V, Rapaka R, Kwon OS, Song BJ. Roles of alcohol and tobacco exposure in the development of hepatocellular carcinoma. Life Sci 2013;92:3-9.
44. Teschke R, Minzlaff M, Oldiges H, Frenzel H. Effect of chronic alcohol consumption on tumor incidence due to dimethylnitrosamine administration. J Cancer Res Clin Oncol 1983;106:58-64.
45. Gellert J, Moreno F, Haydn M, Oldiges H, Frenzel H, et al. Decreased hepatotoxicity of dimethylnitrosamine (DMN) following chronic alcohol consumption. Adv Exp Med Biol 1980;132:237-43.
46. Teschke R. Effect of chronic alcohol pretreatment on the hepatotoxicity elicited by chlorpromazine, paracetamol, and dimethylnitrosamine. Biol Approach Alcohol Natl Inst Alcohol Abuse Alcohol Res Monogr 1983;11:170-89.
48. Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004;34:9-19.
49. Neuman MG, French SW, Zakhari S, Malnick S, Seitz HK, et al. Alcohol, microbiome, life style influence alcohol and non-alcoholic organ damage. Exp Mol Pathol 2017;102:162-80.
50. Osna NA, Donohue TM, Kharbanda KK. Alcoholic liver disease: pathogenesis and current management. Alcohol Res 2017;38:147-61.
51. Basra S, Anand BS. Definition, epidemiology and magnitude of alcoholic hepatitis. World J Hepatol 2011;3:108-13.
53. Teschke R. Alcoholic steatohepatitis (ASH) and acute alcoholic hepatitis (AH): Cascade of events, clinical features, and pharmacotherapy options. Exp Opin Pharmacother 2018;19:779-93.
54. Fleming KM, Withal GP, Card TR, West J. The rate of decompensation and clinical progression of disease in people with cirrhosis: a cohort study. Aliment Pharmacol Ther 2010;32:1343-50.
55. Powell WJ Jr, Klatskin G. Duration of survival in patients with Laennec’s cirrhosis. Influence of alcohol withdrawal, and possible effects of recent changes in general management of the disease. Am J Med 1968;44:406-20.
56. Martini GA, Teschke R. Alcohol abstinence in alcoholic liver disease. Acta Med Scand 1985;703:185-94.
57. O’Grady JG. Liver transplantation alcohol related liver disease: (Deliberately) stirring a hornet’s nest! Gut 2006;55:1529-31.
58. Matsushita H, Takaki A. Alcohol and the hepatocellular carcinoma. BMJ Open Gastroenterol 2019;6:e000260.
59. Ganne-Carrié N, Nahon P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J Hepatol 2019;70:284-93.
60. Schlageter M, Terracciano LM, D’Angelo S, Sorrentino P. Histopathology of hepatocellular carcinoma. World J Gastroenterol 2014;20:15955-64.
61. Schlageter M, Quagliata L, Matter M, Perrina V, Tornillo L, et al. Clinicopathological features and metastatic pattern of hepatocellular carcinoma: an autopsy study of 398 patients. Pathobiology 2016;83:301-7.
62. Mezale D, Strumfa I, Vanags A, Kalva A, Balosis D, et al. Diagnostic algorithm of hepatocellular carcinoma: classics and innovations in radiology and pathology. Intechopen 2018. Available from: https://www.intechopen.com/books/hepatocellular-carcinoma-advances-in-diagnosis-and-treatment/diagnostic-algorithm-of-hepatocellular-carcinoma-classics-and-innovations-in-radiology-and-pathology [last accessed on 7 Nov 2019].
63. Marseglia L, Manti S, D’Angelo G, Nicotera A, Parisi E, et al. Oxidative stress in obesity: a critical component in human diseases. Intl J Mol Sci 2015;16:378-400.
64. Teschke R, Xuan TD. Viewpoint: a contributory role of Shell ginger (Alpinia zerumbet) for human longevity of Okinawa in Japan? Nutrients 2018;10:166.
65. Mendez-Sanchez N, Cruz-Ramon VC, Ramirez-Perez OL, Hwang JP, BarrancoFragoso B, et al. New aspects of lipotoxicity in nonalcoholic steatohepatitis. Int J Mol Sci 2018;19:E2034.
66. Davis GL, Dempster J, Meler JD, Orr DW, Walberg MW, et al. Hepatocellular carcinoma: management of an increasingly common problem. Proc (Bayl Univ Med Cent) 2008;21:266-80.
67. Nahon P, Ganne-Carrié N, Trichet JC, Beaugrand M. Hepatic iron overload and risk of hepatocellular carcinoma in cirrhosis. Gastroenterol Clin Biol 2010;34:1-7.
68. Anstee QM, Daly AK, Day CP. Genetics of alcoholic liver disease. Semin Liver Dis 2015;35:361-74.
69. Donato F, Tagger A, Gelatti U, Parinello G, Boffetta P, et al. Alcohol and hepatocellular carcinoma: the effect of life-time intake and hepatitis virus infections in men and women. Am J Epidemiol 2002;155:323-31.
70. Péquinot G, Tuyns AJ, Berta JL. Ascitic cirrhosis in relation to alcohol consumption. Int J Epidemiol 1978;7:113-20.
71. Tuyns AJ, Péquinot G. Greater risk of ascitic cirrhosis in females in relation to alcohol consumption. Int J Epidemiol 1984;13:53-7.
72. Becker U, Deis A, Sorensen TIA, Gronbaeck M, Borch-Johnsen, et al. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology 1996;23:1025-9.
73. Caballeria J, Frezza M, Hernández-Muñoz R, DiPadova C, Korsten MA, et al. Gastric origin of the first-pass metabolism of ethanol in humans: effect of gastrectomy. Gastroenterology 1989;97:1205-9.
74. Pavlov CS, Casazza G, Semenistaia M, Nikolova D, Tsochatzis E, et al. Ultrasonography for diagnosis of alcoholic cirrhosis in people with alcoholic liver disease. Cochrane Database Syst Rev 2016;3:CD011602.
75. Bartolotta TV, Taibbi A, Midiri M, Lagalla R. Contrast-enhanced ultrasound of hepatocellular carcinoma: where do we stand? Ultrasonography 2019;38:200-14.
77. Cohen JA, Kaplan MM. The SGOT/SGPT ratio an indicator of alcoholic liver disease. Dig Dis Sci 1979;24:835-8.
78. Teschke R, Neuefeind M, Nishimura M, Strohmeyer G. Hepatic gamma-glutamyltransferase activity in alcoholic fatty liver: comparison with other liver enzymes in man and rats. Gut 1983;24:625-30.
79. Heckley GA, Jarl J, Asamoah BO, G-Gerdtham U. How the risk of liver cancer changes after alcohol cessation: a review and meta-analysis of the current literature. BMC Cancer 2011;11:446.
80. Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, et al; Global Burden of Disease Liver Cancer Collaboration. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the global burden of disease study 2015. JAMA Oncol 2017;3:1683-91.
Cite This Article

How to Cite
Teschke, R. Hepatocellular carcinoma in alcoholic liver disease: mechanistic considerations and clinical facts. Hepatoma. Res. 2019, 5, 40. http://dx.doi.org/10.20517/2394-5079.2019.0017
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.