



Electro-assisted assembly of conductive polymer and soft hydrogel into core-shell hybrids


Abstract
Soft hydrogels have become an important class of materials for mimicking and interfacing biological soft tissues with potential applications in drug delivery, tissue engineering and bioelectronics. Creative methods for integrating hydrogels with other materials such as organic conductors are highly desired. Here, we describe the single-step electrosynthesis of PEDOT/alginate into core-shell hybrid structures via an electrochemical-chemical-chemical mechanism. Using a pulsed electropolymerisation protocol, we generated PEDOT in either oxidized or reduced form. By-products of this electrochemical step trigger the chemical reactions for the concomitant assembly of alginate hydrogels. Characterization evidences that PEDOT (core) and alginate (shell) compartments form an electrochemically integrated interface. During growth, both can be loaded with useful cargo. We loaded a negatively charged small molecule and investigated passive and electroactive release mechanisms from the two compartments. Our electro-assisted assembly/crosslinking of integrated PEDOT/alginate hybrids contributes a promising approach to the design of functional interfaces for applications in controlled release and soft electronics.
Keywords
INTRODUCTION
Hydrogels are crosslinked polymer networks swollen with water (typically > 95%wt). They can be built from a wide range of natural and synthetic macromolecules. As bioengineering materials, they offer useful properties such as tissue-like viscoelasticity, tuneable degradation, three-dimensional matrix for supporting cell growth and sequestration or release of biomolecules, among others[1,2]. A notable example is alginate (extracted from brown algae) which consists of the anionic polysaccharide alginic acid that can be crosslinked by metal ions such as Ca2+. The resultant network readily absorbs water and swells to form a hydrogel with a pore size of a few nanometers[3]. Alginate hydrogels are used in clinical devices such as wound dressings and implant coatings, as drug delivery microparticles or as cell encapsulates in bioprinting technologies[4,5].
Hydrogels can be composed of more than one polymer network to enable hybrid functionalities. For example, the incorporation of an interpenetrating conductive polymer network results in hydrogels with mixed mode (electronic-ionic) conductivity[6]. This is desirable for bioelectronic interfacing technologies where the hydrogel can enhance the charge injection capacity of electrodes, serve as a mechanical buffer between rigid substrates and tissue, improve cell adhesion, or facilitate drug release[7-10]. A notable example is poly (3,4-ethylenedioxythiophene) (PEDOT) which has good electroactivity and processability when combined with a polyanion dopant[10-16]. Although PEDOT is mostly processed in thin-film coatings, recent studies have integrated it into conductive hydrogels[17-22]. For instance, PEDOT and alginate are often reported as composites or blends attempting to merge their constituent properties in the resulting material[23-28].
Hydrogels can be formed by various mechanisms, including ionic or covalent crosslinks, as well as interpenetrating polymer networks[29]. Although the electrostatic interaction is stronger and more stable in nature (e.g., minerals, rocks), due to the high water content in hydrogels, the resulting ionic crosslinks generate typically brittle materials with poor mechanical stability, as is the case with most alginate based hydrogels[30]. This makes integration with other materials (where soft-rigid interfaces are present) challenging and may limit some practical applications[31]. This is even more pressing in the case of conductive hydrogels, where the interface with an organic semiconductor or a metallic conductor additionally requires efficient charge exchange[32,33]. Thus, despite remarkable progress where conductive hydrogels have achieved extreme stretchability, softness and conductivity, relatively little has been done to engineer good interfaces to other materials as will be needed in practical systems.
One option is to utilise surface functionalisation or treatments (e.g., silanization) that improve adhesion via covalent and/or ionic bonds[31,33,34]. These methods are effective; however, they require additional steps and add complexity to the processing of hybrid structures. As an alternative, electrodeposition methods can offer a simple strategy to facilitate the direct growth of soft materials, especially over conductive substrates, controlling electron transfer and adhesion process at the interface[32,35-38]. A number of promising electrochemical approaches have already been reported. For instance, water hydrolysis or redox mediators have been used to promote the electrogelation of a range of ionically crosslinked biomatrixes[39-44]. Copper ions electrochemically released from a sacrificial layer have been shown to coordinate PEDOT:PSS microparticles and the formation of conductive hydrogels[32]. Recently, we have proposed an electro-assisted method for forming hydrogels on conductive surfaces using an electrochemical-chemical-chemical (ECC) mechanism. This includes deposition of conductive hybrid PEDOT/alginate films[36]. However, application of traditional electrodeposition protocols highlighted several limitations, such as slow growth kinetics and low levels of hydration of the final material.
In the present work, we propose an electro-assisted assembly of core-shell structures formed from PEDOT (core) and alginate hydrogel (shell). This is achieved via pulsed electrochemical protocols and the ECC mechanism, which results in significant improvement of growth kinetics of the core and shell compartments. We find that PEDOT and the alginate hydrogel form a well integrated electrochemical and mechanical interface. We explore the core-shell structures as passive/sensing release and electrically controlled delivery vehicles. In our system, the hydrogel is formed on a robust conductive substrate which improves the overall integration. This is in contrast with freestanding structures that may be difficult to handle. The strength of our approach is that even such brittle and delicate hydrogels (such as alginate) can be integrated into a device. In terms of clinical application, we envisage the hydrogels as functional coatings on electrodes that can transform a traditional electrode into a depot for drug or cell delivery.
METHODS
Materials. The chemical reagents 3,4-ethylenedioxythiophene (EDOT), sodium alginate, calcium carbonate, sodium dodecyl sulphate (SDS), fluorescein and sodium citrate were purchased from Sigma-Aldrich. All solutions were prepared with deionized Milli-Q water (18.2 MΩ) or with phosphate saline buffer (PBS)
Electrodeposition solution. The solution composition was adapted from a previous work[36]. Firstly,
Electrochemical measurements. Cyclic voltammetry (CV), chronoamperometry (CA) and electrochemical impedance spectroscopy (EIS) were performed using a potentiostat/galvanostat (PARSTAT3000, AMETEK) controlled using VersaStudio 2.60.2 software. Pulsed protocol methods were programmed a “Loop” (number of desired pulses) containing CA at electrodeposition potential (+1.4 V) for 0.5 s plus either OCP (for OCPI protocol) or CA at 0 V (for RPI protocol) for 10 s. CV traces were recorded in PBS as supporting electrolytes from -1.2 V to +0.8 V at a scan rate of 100 or 50 mV s-1. EIS data were recorded from 1 MHz to 0.1 Hz, with an excitation amplitude of 10 mV (RMS) at 10 points per decade. The working electrode employed was gold wire with submersed lengths ranging from 1.27 to 2.54 cm. A commercially available Ag/AgCl/KCl 3M (BASi) electrode and a platinum coil were used as reference and counter electrodes, respectively. For statistical analysis, all experiments were made in triplicate using three different gold wire electrodes and freshly prepared solutions. Unless stated otherwise, data is reported as the mean ± standard deviation.
All charges were calculated by Q = I × t, where I is the current density (A mm-2) and t is time (seconds). Parameter fitting and circuit simulation were conducted using the NOVA 2.1 software (Metrohm Autolab). Resistance was calculated from the constant phase element (CPE) using the following equation[45,46]:

where R is resistance, Y0 is the pseudocapacitance constant value, 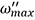 is the frequency where the imaginary part is highest and n is the deviation from ideal capacitor (n = 0 is a pure resistor, n = 1 is a pure capacitor).
is the frequency where the imaginary part is highest and n is the deviation from ideal capacitor (n = 0 is a pure resistor, n = 1 is a pure capacitor).
Fluorescence quantification. To quantify the concentration of the fluorescein molecule, a florescence spectrometer was used to measure light intensity. A reference curve was prepared using serial dilutions from 1 mg mL-1 to 1.5 10-5 mg mL-1. Molecule excitation was at 485 nm and emission was measured at 528 nm. The concentration of fluorescein was calculated using the reference curve using GraphPad.
Scanning electron microscopy - energy dispersive x-ray spectroscopy (SEM-EDS). A Philips/FEI XL-20 SEM (Philips, UK) scanning electron microscope was used to image dehydrated hydrogel samples. Cross section was obtained by cutting the hydrogels using a razor blade when freshly prepared. The samples were dehydrated by replacing water with ethanol and were processed in a critical point dryer (CPD). Samples were sputter coated with gold before SEM imaging. The gold-coated hydrogels were then imaged with an accelerating voltage of 15 kV.
RESULTS
Conductive Hybrid PEDOT/alginate Hydrogels. We investigated a number of electrochemical strategies to generate PEDOT/alginate hybrid hydrogels. Firstly, we attempted to use water hydrolysis (+1.85 to +2.0 V) to promote gelation of alginate in parallel with electropolymerisation of PEDOT, which also occurs in this potential window. This did not produce a core-shell structure but a hybrid hydrogel with blue colour and gel aspect [Supplementary Figure 1]. The resultant structure has poor electroactivity, likely because gas produced during water hydrolysis is removing PEDOT from the gold surface, and hence the hybrid hydrogel is electrically not well-connected to the substrate (no electroactivity). We then turned to an alternative strategy where the protons generated during EDOT polymerisation are used to promote the gelation of alginate. We added the surfactant SDS (70 mM) to the aqueous polymerisation solution to increase the concentration of EDOT monomers to 50 mM[47] which is expected to dramatically increase the availability of protons. This resulted in the growth of a core-shell structure with a significant amount of alginate hydrogel layer (shell) which enveloped a film of PEDOT (core) with fractal morphology [Figure 1]. Here SDS (an anionic surfactant) is chosen over other surfactants because it is compatible with the negative charges on the alginate molecule. As illustrated in Supplementary Figure 2, SDS does not precipitate the alginate solution, which means that the surfactant itself does not interfere with the electrically assisted gelation processes.

Figure 1. Electrodeposition of core-shell structures. (A) Chemical reactions involved in electropolymerisation (1) of EDOT to neutral state of PEDOT (benzoid structure, top) and redox equilibrium (blue block) to oxidized PEDOT (quinoid structure, bipolaron represented in blue, bottom) followed by (2) decomposition of calcium carbonate; and (3) complexation with alginate macromolecules representing the alginate hydrogel formation (green block). (B) Schematic representation of the 3-electrode electrochemical cell with reference electrode (RE), working electrode (WE) and counter electrode (CE). On the left, coaxial representation (red dashed square) of the core-shell structures deposited over gold wire. On the right, schematic representation (blue) of molecules and ionic species present in the bulk electrolyte, above the critical micellar concentration (CMC) and WE potential of +1.4 V (vs. Ag/AgCl/KCl 3M). The respective order of chemical reactions (1-3) leading to core-shell formation is indicated. (C) Photograph of the PEDOT/alginate structures electrodeposited over a gold wire, followed by SEM images of the alginate shell surface (middle) and PEDOT core layer (right). (D) SEM image (left) of a cross section at the PEDOT/alginate interface and atomic distribution (right) of sulphur (S, Kα1 in red) and calcium (Ca, Kα1 in green).
The reaction follows an electrochemical-chemical-chemical mechanism (ECC). At +1.4 V (vs.
Growth Kinetics of Core-Shell Hydrogels. A typical electrodeposition method is the step potential, as illustrated in Figure 2A(i) (constant electric potential applied for a pre-set time). Under this condition, the electropolymerisation current rapidly drops when the initial monomer concentration is depleted at the electrode’s interface. Later, it reaches an equilibrium with new monomers diffusing from the bulk solution

Figure 2. Growth kinetics of PEDOT/alginate structures. (A) Program for electrodeposition in (i) traditional step electric potential (electrodeposition in blue), (ii) pulses of 0.5 s (blue) followed by holding at OCP (green) for 10 s, called open circuit potential interpulse (OCPI) protocol and (iii) pulses of 0.5 s (blue) followed by holding at 0 V (reduction potential, orange), called reducing potential interpulse (RPI) protocol. (B) Charge versus number of polymerisation pulses, discriminating the total oxidation charge (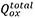 , black), the total reduction charge (
, black), the total reduction charge (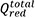 , red) and the calculated actual electrodeposition charge (Qelectrodeposition, green) for the RPI protocol. (C) Kinetics of the PEDOT growth (produced using the OCPI protocol) showing zero order growth from 15 to 1000 polymerisation pulses. (D) Kinetics of the alginate hydrogel growth (produced using the OCPI protocol) showing zero order growth from 15 to 120 polymerisation pulses. (E) Photograph of the PEDOT/alginate hydrogel electrodeposited over a gold wire (13 mm length) evidencing the PEDOT (core) and alginate (shell) rich zones. (F) Growth profiles of PEDOT and alginate vs. pulse number using the OCPI protocol. Inset: Cross section scheme of the formed structure indicating the gold wire (diameter 0.1 mm), PEDOT and alginate diameters used to calculate the respective volumes.
, red) and the calculated actual electrodeposition charge (Qelectrodeposition, green) for the RPI protocol. (C) Kinetics of the PEDOT growth (produced using the OCPI protocol) showing zero order growth from 15 to 1000 polymerisation pulses. (D) Kinetics of the alginate hydrogel growth (produced using the OCPI protocol) showing zero order growth from 15 to 120 polymerisation pulses. (E) Photograph of the PEDOT/alginate hydrogel electrodeposited over a gold wire (13 mm length) evidencing the PEDOT (core) and alginate (shell) rich zones. (F) Growth profiles of PEDOT and alginate vs. pulse number using the OCPI protocol. Inset: Cross section scheme of the formed structure indicating the gold wire (diameter 0.1 mm), PEDOT and alginate diameters used to calculate the respective volumes.
We next define the following charges: the and as the total charges applied in each pulse for oxidation and reduction processes, respectively. Qelectropolymerization is the charge that directly contributes to polymerisation by linking EDOT monomers, forming benzoid structure [Figure 1A]. Qox PEDOT is the charge used to oxidize PEDOT chains (obtaining quinoid structure, Figure 1A) after the polymer is already formed. We assume that the charge delivered at positive electrode potentials, contributes to both Qelectropolymerization and Qox PEDOT. Similarly, Qred PEDOT is the charge expended to reduce the PEDOT from quinoid to benzoid structure. Since already formed PEDOT cannot depolymerize, charges flowing during the interpulse can only contribute to , thus = Qred PEDOT. Finally, Q is the summation of all charges at the end of a pulse or sequence of pulses [Figure 2B]; we can thus write:
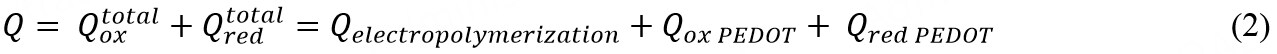
For RPI, after a time (here empirically determined to be 60 pulses), a state where Qox PEDOT = -Qred PEDOT is reached. This is because, during the reducing interpulse period (electrode potential = 0 V), the current quickly drops to zero, suggesting PEDOT is left in a neutral state [Supplementary Figures 7 and 8]. This allows us to obtain Qelectropolymerization. For the OCPI protocol, we electrodeposited the hybrid hydrogel first with a pulse train and afterwards applied a long reducing pulse (0 V for 20 min) to obtain the condition where
The estimate for Qelectropolymerization, therefore, allows us to determine the effective surface coverage (Г) which quantifies the amount of polymer deposited per unit area of electrode[49].

where F is the Faraday constant (96,486 C mol-1), n is the number of electrons transferred in the electrodeposition process (n = 1, for linking two EDOT monomers) and A is the geometric area of the electrode (gold wire). Knowing Qelectropolymerization, we also estimated the number of generated protons (reaction in Figure 1) and therefore the number of moles of Ca2+ released. Assuming that one Ca2+ is sufficient to coordinate the crosslinking of two alginate molecules, we can link the (electron) charge to the amount of alginate hydrogel produced in the shell. From the reactions described in Figure 1, we can write the following stoichiometric relationship of electron:proton:calcium:alginate as 1:1:½:1 respectively. This allowed us to obtain the kinetics of growth for both PEDOT and alginate components obtained via the OCPI protocol. As illustrated in Figure 2C, the growth of the PEDOT core follows order zero with 0.25 ± 0.01 µmol cm-2 pulse-1 (r = 0.9983) for all polymerisation pulses applied. Figure 2D shows that the growth of the alginate shell follows order zero as 0.07 ± 0.01 µmol mm-3 pulse-1 (r = 0.9997) until 120 polymerisation pulses. After 120 pulses, the zero order growth kinetics breaks down. Figure 2E shows a picture of the PEDOT/alginate structure obtained with 240 pulses (OCPI), highlighting the interface of PEDOT (core, blue) and alginate (shell, green) rich zones. Figure 2F evidences the growth profile of the core and shell based on diameter increase. The diameters increase with a similar trend as discussed above using kinetics parameters [Figure 2D]. The growth profile of the alginate hydrogel presented here is similar to pure alginate formed over electrodes, tending to reach a plateau for long electrodeposition periods[36].
Electrochemical properties of the PEDOT-Alginate interface. To investigate if the core and shell layers are electrochemically linked, we performed cyclic voltammetry and impedance spectroscopy measurements. Figure 3A shows normalized cyclic voltammograms of bare gold wire (black), PEDOT:SDS (without alginate, grey) and the PEDOT/alginate structures electrodeposited with step potential (1000 s, red), RPI

Figure 3. Electrochemistry of the PEDOT/alginate interface. (A) Cyclic voltammograms (normalised by area and Qelectrodeposition) for bare gold wire (black), PEDOT:SDS (control without alginate, grey) and PEDOT/alginate structures electrodeposited with step potential (red), pulsed RPI protocol (green) and OCPI protocol (blue) in PBS (pH = 7.4) as supporting electrolyte and scan rate of 50 mV s-1.
Electrochemical impedance spectroscopy (EIS) for the various deposition protocols is shown in Figure 3B. Again, the structures electrodeposited using the two pulsed methods presented lower impedance when compared to PEDOT:SDS and PEDOT/alginate (step). Between the structures obtained via pulsed methods, the OCPI protocol shows a reduced total impedance, especially for lower frequencies.
The charge storage capacity (CSC) for bare gold wire (black), PEDOT:SDS (without alginate, grey) and the core-shell structures electrodeposited with step potential (1000 s, red), RPI (60 pulses, green) and OCPI
The behaviour of core-shell structures was also investigated by fitting an equivalent circuit model to EIS measurements [Supplementary Figure 11]. Figure 3D shows our proposed equivalent circuit (top) and a schematic representation of the core-shell structures (bottom). Assuming the interface between PEDOT and the alginate hydrogel is electrically integrated, the PEDOT should correspond to a constant phase element (CPE) that is both electronically and ionically conductive and connected in series with a capacitance (C) representing the ionically conductive alginate hydrogel. The supporting electrolyte (PBS) is modelled by a resistance (Rs). Figure 3E shows a table containing the values of the components extracted by fitting EIS spectra. We note a relatively low value for the CPE exponent (n) for the PEDOT element (0.274 and 0.172 for OCPI and RPI protocols, respectively). It suggests that here PEDOT acts more as a resistor (electronically conductive) than a capacitor. Resistor values (RPEDOT) of 0.168 and 0.070 Ω cm2 for OCPI and RPI protocols were extracted, respectively (see methods section). The capacitance (C) was found to be 22.4 and 6.54 mF for OCPI and RPI protocols, respectively. These observations agree with our previous characterizations [Figure 2] where the interface produced using OCPI protocol is fully doped (i.e., loaded with charges), while the interface produced using RPI protocol is fully dedoped (i.e., neutral). In addition, the major contribution to the capacitance (C) should come from a thicker alginate layer. A strong influence of the initial doping state of PEDOT on the capacitance of the alginate compartment suggests good integration between the two materials.
The electrochemical properties of a number of PEDOT/alginate materials reported in the literature are presented in Table 1. The current densities supported by the reported materials range from 5 µA cm-2 to
Comparison between CVs and PEDOT resistance for PEDOT/Alginate materials
| Name | Method | Current density | RPEDOT& | Reference |
| PEDOT/Alginate hydrogel | PEDOT:PSS in Alginate matrix | ~8 µA cm-2, 100 mM CaCl2 | 62.9 kΩ | Garcia-Torres, |
| PEDOT:PSS/Alginate | Microelectrode array, PEDOT:PSS mixed in Alginate matrix | ~0.8 µA*, PBS | 5-30 kΩ# | Ghezzi, et al.[16] |
| Alginate-PCNT-PEDOT/PSS hydrogel | Microwire, PEDOT:PSS coated MWCNT in Alginate hydrogel | ~1 mA cm-2, 9% NaCl | 123 kΩ | Wang, et al.[23] |
| PEDOT/hydrogel/BDNF-coated implants | Pt/Ir cochlear implant (microelectrode), electrodeposition of PEDOT:PSS | ~3 µA*, PBS | - | Chikar, et al.[24] |
| PEDOT7Alg3 | PEDOT/Alginate scaffolds | 300 µA*, not informed | - | Yang, et al.[25] |
| Hybrid PEDOT/Alginate hydrogel | Macroelectrode (centimeter size) electrodeposition, pulsed method | 60 mA cm-2, PBS | 0.168 Ω cm2 (2.1 Ω) | Present work |
Loading and Release of a negatively charged small molecule. We used fluorescein (widely known as model negatively charged small molecule[50-52]) to explore the potential of core-shell structures as encapsulation and delivery system. To encapsulate the target molecule in both the PEDOT (core) and alginate (shell) compartments in a single step, we added 1 mg mL-1 of fluorescein to the electrodeposition solution [Figure 4]. Our approach differs from previously reported work where the hydrogel is prepared first and then loaded either passively or by oxidizing the conducting polymer chain in the presence of the cargo molecule (and other ions as well)[53-56]. Figure 4A and B show a schematic representation and a photograph of the structures loaded with fluorescein.

Figure 4. Passive/sensing release and electroactive controlled release of fluorescein. (A) Schematic representation of the core-shell structure. Shell compartment: passive release from the alginate layer. Core compartment: electroactive controlled release from PEDOT (quinoid structure, bottom). (B) Photograph of the core-shell structure loaded with fluorescein. (C) Concentration of fluorescein encapsulated in the volume of the alginate shell. The horizontal dashed line (red) indicates the fluorescein concentration in the polymerisation solution (1 mg mL-1). (D) Relationship between the amount of fluorescein encapsulated in the shell and the polymerisation charge applied to form the core-shell structure. Statistical analyses for (C and D) were made in triplicate over three different gold wire electrodes. Statistical significance was determined using one-way analysis of variance (ANOVA)/Tukey correction, n.s. is non-significant (P > 0.05), *P < 0.05 and **P < 0.001. (E) Passive release of fluorescein quantified by fluorescence (green, left axis) and total impedance change of the interface (blue, right axis). (F) Amount of fluorescein actively released from the core at different electric potentials. Each release experiment lasts 40 min. (G) Relationship between the amount of fluorescein encapsulated in the core and the polymerisation charge applied for core-shell structure formation. (H) Electroactive release of fluorescein under controlled conditions: ON (black, -1.2 V), OFF (green, +0.8 V), passive (red, OCP), P30 (blue, pulse for 30 s at -1.2 V), P8 (purple, pulse for 8 s at
Our integrated interface consists of two distinct compartments: (1) the alginate shell capable of passively releasing the target molecule (top, green); and (2) the PEDOT core capable of electrically controlled release (bottom, blue). As the cargo molecule is negatively charged, the OCPI protocol was employed here to generate oxidized PEDOT (positively charged). The amount of loaded fluorescein can be inferred by fully releasing the cargo from both compartments. Figure 4C shows the concentration of fluorescein embedded in the alginate volume (i.e., excluding any fluorescein embedded in PEDOT core). This was obtained by completely dissolving the alginate layer with sodium citrate (50 mM). Interestingly, for the hybrid gel produced with 120 pulses, the concentration of encapsulated fluorescein is higher than the concentration in the bulk solution (1 mg mL-1, red dotted line). This is likely due to the SDS micelles assisting in concentrating the target molecules. Initially, the concentration increases linearly with pulse number following the growth kinetics of the shell. Beyond 120 pulses, the concentration decreases, likely because of the change in growth kinetics previously discussed [Figure 2]. Figure 4D shows the fluorescein amount passively released (gel submerged in PBS for 24 h) from core-shell structures formed with different amounts of charge/pulse number. Within experimental error, we observe a constant amount of released fluorescein per total charge unit () (no significant difference P > 0.005). Figure 4E shows the temporal profile of fluorescein release from the shell compartment (green, left axis) and the evolution of the interface impedance (modulus, at 1 Hz). Although other effects cannot be ruled out, it appears that the decrease in impedance correlates with the release of fluorescein and can thus be used to monitor the process.
For studying electroactive release from the PEDOT core, the alginate layer was first dissolved in sodium citrate. Figure 4F shows the electroactive release of fluorescein under different electric potentials (within the aqueous electrochemical window) applied for 40 min in PBS media. The lower the electric potential applied, the higher the amount of fluorescein released. Additionally, it is worth mentioning that the amount of fluorescein stored in the PEDOT core compartment (and available for controlled release) is roughly 10 times lower than that in the alginate shell (passive release) compartment. Figure 4G shows the total amount of fluorescein electrically released as function of the charge supplied for core-shell structure formation ).
CONCLUSIONS
In this work, we describe single-step electrosynthesis of conductive core-shell structures of PEDOT and alginate hydrogel. For this, we developed pulsed protocols to enhance the growth of the alginate compartment. Charge considerations enable us to determine the amount of electrodeposited materials, and their growth kinetics. Electrochemical characterization revealed that the interface is highly electroactive, indicating good integration between the PEDOT and alginate compartments. We loaded the structures with a negatively charged model molecule for demonstration of passive release (and its monitoring) and controlled electroactive release from the alginate and PEDOT compartments, respectively. Our electro-assisted assembly and loading approach may contribute to the design of electrical hydrogel devices for applications in biointerfaces and soft electronics. For instance, in the context of implantable electrode arrays, the alginate shell compartment may enable burst release of a high dose of a drug (e.g., the anti-inflammatory drug dexamethasone), loaded vesicles or encapsulated cells, while the PEDOT core compartment can provide sustained long-term maintenance dosing.
DECLARATIONS
Authors’ contributionsConceived the idea: Da Silva AC, Minev IR
Planned and performed the experiments: Da Silva AC
Performed SEM analysis and fluorescence quantification: Paterson TE
Analysed results and wrote the manuscript: Da Silva AC, Paterson TE, Minev IR
Availability of data and materialsNot applicable.
Financial support and sponsorshipAll authors acknowledge funding from ERC Starting Grant: IntegraBrain (804005). We acknowledge assistance from Christopher Hill, Department of Molecular Biology and Biotechnology for SEM-EDS and CPD analysis.
Conflicts of InterestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
Supplementary MaterialsREFERENCES
1. Tringides CM, Boulingre M, Khalil A, Lungjangwa T, Jaenisch R, Mooney DJ. Tunable conductive hydrogel scaffolds for neural cell differentiation. Adv Healthc Mater 2022:e2202221.
2. Freudenberg U, Atallah P, Limasale YDP, Werner C. Charge-tuning of glycosaminoglycan-based hydrogels to program cytokine sequestration. Faraday Discuss 2019;219:244-51.
3. Simpliciano C, Clark L, Asi B, et al. Cross-linked alginate film pore size determination using atomic force microscopy and validation using diffusivity determinations. J Surf Eng Mater Adv Technol 2013;03:1-12.
4. Rastogi P, Kandasubramanian B. Review of alginate-based hydrogel bioprinting for application in tissue engineering. Biofabrication 2019;11:042001.
5. Correa S, Grosskopf AK, Lopez Hernandez H, et al. Translational applications of hydrogels. Chem Rev 2021;121:11385-457.
6. Paulsen BD, Fabiano S, Rivnay J. Mixed ionic-electronic transport in polymers. Annu Rev Mater Res 2021;51:73-99.
7. Rong Q, Lei W, Liu M. Conductive hydrogels as smart materials for flexible electronic devices. Chemistry 2018;24:16930-43.
8. Distler T, Boccaccini AR. 3D printing of electrically conductive hydrogels for tissue engineering and biosensors - A review. Acta Biomater 2020;101:1-13.
9. Shur M, Fallegger F, Pirondini E, et al. Soft printable electrode coating for neural interfaces. ACS Appl Bio Mater 2020;3:4388-97.
10. Tondera C, Akbar TF, Thomas AK, et al. Highly conductive, stretchable, and cell-adhesive hydrogel by nanoclay doping. Small 2019;15:e1901406.
11. Bhat MA, Rather RA, Shalla AH. PEDOT and PEDOT:PSS conducting polymeric hydrogels: a report on their emerging applications. Synth Met 2021;273:116709.
12. Boehler C, Aqrawe Z, Asplund M. Applications of PEDOT in bioelectronic medicine. Bioelectron Med 2019;2:89-99.
13. Heo DN, Lee SJ, Timsina R, Qiu X, Castro NJ, Zhang LG. Development of 3D printable conductive hydrogel with crystallized PEDOT:PSS for neural tissue engineering. Mater Sci Eng C 2019;99:582-90.
14. Fu F, Wang J, Yu J. Interpenetrating PAA-PEDOT conductive hydrogels for flexible skin sensors. J Mater Chem C 2021;9:11794-800.
15. Lacour SP, Courtine G, Guck J. Materials and technologies for soft implantable neuroprostheses. Nat Rev Mater 2016;1:16063.
16. Ferlauto L, D’Angelo AN, Vagni P, et al. Development and characterization of PEDOT:PSS/alginate soft microelectrodes for application in neuroprosthetics. Front Neurosci 2018;12:648.
17. Shi H, Liu C, Jiang Q, Xu J. Effective approaches to improve the electrical conductivity of PEDOT:PSS: a review. Adv Electron Mater 2015;1:1500017.
18. Liu Y, Liu J, Chen S, et al. Soft and elastic hydrogel-based microelectronics for localized low-voltage neuromodulation. Nat Biomed Eng 2019;3:58-68.
20. Li G, Huang K, Deng J, et al. Highly conducting and stretchable double-network hydrogel for soft bioelectronics. Adv Mater 2022;34:e2200261.
22. Cheng T, Zhang Y, Wang S, et al. Conductive hydrogel-based electrodes and electrolytes for stretchable and self-healable supercapacitors. Adv Funct Mater 2021;31:2101303.
23. Wang K, Tian L, Wang T, et al. Electrodeposition of alginate with PEDOT/PSS coated MWCNTs to make an interpenetrating conducting hydrogel for neural interface. Compos Interfaces 2019;26:27-40.
24. Chikar JA, Hendricks JL, Richardson-Burns SM, Raphael Y, Pfingst BE, Martin DC. The use of a dual PEDOT and RGD-functionalized alginate hydrogel coating to provide sustained drug delivery and improved cochlear implant function. Biomaterials 2012;33:1982-90.
25. Yang B, Yao F, Ye L, et al. A conductive PEDOT/alginate porous scaffold as a platform to modulate the biological behaviors of brown adipose-derived stem cells. Biomater Sci 2020;8:3173-85.
26. Paradee N, Sirivat A. Electrically controlled release of benzoic acid from poly(3,4-ethylenedioxythiophene)/alginate matrix: effect of conductive poly(3,4-ethylenedioxythiophene) morphology. J Phys Chem B 2014;118:9263-71.
27. Babeli I, Ruano G, Puiggalí-jou A, Ginebra M, Alemán C, Garcia-torres J. Self-healable and eco-friendly hydrogels for flexible supercapacitors. Adv Sustain Syst 2021;5:2000273.
28. Guo J, Yu Y, Wang H, Zhang H, Zhang X, Zhao Y. Conductive polymer hydrogel microfibers from multiflow microfluidics. Small 2019;15:e1805162.
29. Madduma-bandarage USK, Madihally SV. Synthetic hydrogels: synthesis, novel trends, and applications. J Appl Polym Sci 2021;138:50376.
30. Ji D, Park JM, Oh MS, et al. Superstrong, superstiff, and conductive alginate hydrogels. Nat Commun 2022;13:3019.
31. Yuk H, Zhang T, Lin S, Parada GA, Zhao X. Tough bonding of hydrogels to diverse non-porous surfaces. Nat Mater 2016;15:190-6.
32. Feig VR, Tran H, Lee M, et al. An Electrochemical gelation method for patterning conductive PEDOT:PSS hydrogels. Adv Mater 2019;31:e1902869.
33. Inoue A, Yuk H, Lu B, Zhao X. Strong adhesion of wet conducting polymers on diverse substrates. Sci Adv 2020;6:eaay5394.
34. Yang J, Bai R, Chen B, Suo Z. Hydrogel adhesion: a supramolecular synergy of chemistry, topology, and mechanics. Adv Funct Mater 2020;30:1901693.
35. Azmi S, Frackowiak E. Redox activity from the electrolyte and electrode in electrochemical capacitors. Electrochem Commun 2022;138:107289.
36. Silva AC, Wang J, Minev IR. Electro-assisted printing of soft hydrogels via controlled electrochemical reactions. Nat Commun 2022;13:1353.
37. Silva AC, Akbar TF, Paterson TE, Werner C, Tondera C, Minev IR. Electrically controlled click-chemistry for assembly of bioactive hydrogels on diverse micro- and flexible electrodes. Macromol Rapid Commun 2022;43:e2200557.
38. Fernandes R, Wu L, Chen T, et al. Electrochemically induced deposition of a polysaccharide hydrogel onto a patterned surface. Langmuir 2003;19:4058-62.
39. Cheng Y, Luo X, Betz J, et al. In situ quantitative visualization and characterization of chitosan electrodeposition with paired sidewall electrodes. Soft Matter 2010;6:3177.
40. Gray KM, Liba BD, Wang Y, et al. Electrodeposition of a biopolymeric hydrogel: potential for one-step protein electroaddressing. Biomacromolecules 2012;13:1181-9.
41. Cheng Y, Luo X, Betz J, Payne GF, Bentley WE, Rubloff GW. Mechanism of anodic electrodeposition of calcium alginate. Soft Matter 2011;7:5677.
42. Kim E, Xiong Y, Cheng Y, et al. Chitosan to connect biology to electronics: fabricating the bio-device interface and communicating across this interface. Polymers 2015;7:1-46.
43. Liu Y, Zhang B, Gray KM, et al. Electrodeposition of a weak polyelectrolyte hydrogel: remarkable effects of salt on kinetics, structure and properties. Soft Matter 2013;9:2703.
44. Li J, Kim E, Gray KM, et al. Mediated electrochemistry to mimic biology’s oxidative assembly of functional matrices. Adv Funct Mater 2020;30:2001776.
45. Córdoba-torres P. Relationship between constant-phase element (CPE) parameters and physical properties of films with a distributed resistivity. Electrochim Acta 2017;225:592-604.
46. Hirschorn B, Orazem ME, Tribollet B, et al. Determination of effective capacitance and film thickness from constant-phase-element parameters. Electrochim Acta 2010;55:6218-27.
47. Sakmeche N, Aaron JJ, Fall M, et al. Anionic micelles; a new aqueous medium for electropolymerization of poly(3,4-ethylenedioxythiophene) films on Pt electrodes. Chem Commun 1996:2723.
49. Hutton LA, Vidotti M, Patel AN, Newton ME, Unwin PR, Macpherson JV. Electrodeposition of nickel hydroxide nanoparticles on boron-doped diamond electrodes for oxidative electrocatalysis. J Phys Chem C 2011;115:1649-58.
50. Kleber C, Lienkamp K, Rühe J, Asplund M. Electrochemically Controlled drug release from a conducting polymer hydrogel (PDMAAp/PEDOT) for local therapy and bioelectronics. Adv Healthc Mater 2019;8:e1801488.
51. Bazylevich A, Patsenker LD, Gellerman G. Exploiting fluorescein based drug conjugates for fluorescent monitoring in drug delivery. Dye Pigment 2017;139:460-72.
52. Jamwal HS, Chauhan GS. Designing silica-based hybrid polymers and their application in the loading and release of fluorescein as a model drug and diagnostic agent. Adv Polym Technol 2018;37:411-8.
53. Bagheri B, Zarrintaj P, Surwase SS, et al. Self-gelling electroactive hydrogels based on chitosan-aniline oligomers/agarose for neural tissue engineering with on-demand drug release. Colloids Surf B Biointerfaces 2019;184:110549.
54. Liang Y, Zhao X, Hu T, et al. Adhesive hemostatic conducting injectable composite hydrogels with sustained drug release and photothermal antibacterial activity to promote full-thickness skin regeneration during wound healing. Small 2019;15:e1900046.
55. Qu J, Liang Y, Shi M, Guo B, Gao Y, Yin Z. Biocompatible conductive hydrogels based on dextran and aniline trimer as electro-responsive drug delivery system for localized drug release. Int J Biol Macromol 2019;140:255-64.
56. Qu J, Zhao X, Ma PX, Guo B. Injectable antibacterial conductive hydrogels with dual response to an electric field and pH for localized “smart” drug release. Acta Biomater 2018;72:55-69.
57. Bansal M, Dravid A, Aqrawe Z, Montgomery J, Wu Z, Svirskis D. Conducting polymer hydrogels for electrically responsive drug delivery. J Control Release 2020;328:192-209.
58. Park Y, Jung J, Chang M. Research progress on conducting polymer-based biomedical applications. Appl Sci 2019;9:1070.
59. Cao Y, Samy KE, Bernards DA, Desai TA. Recent advances in intraocular sustained-release drug delivery devices. Drug Discov Today 2019;24:1694-700.
Cite This Article
 , ... Ivan Rusev Minev
, ... Ivan Rusev MinevHow to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright















Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].