


Duchenne muscular dystrophy, one of the most complicated diseases for gene therapy

Abstract
Gene therapy for Duchenne muscular dystrophy (DMD) is hindered by many pitfalls related in particular to the limitations of current technologies, the specificities of muscle and cardiac targets, and the disease itself, a chronic, multisystem, dystrophic and inflammatory disorder. Following RNA-based therapies, DNA gene transfer, mainly based on adeno-associated viral vectors, is now able to deliver therapeutic genetic sequences on a massive scale, and the first antisense and adeno-associated virus (AAV)-microdystrophin gene products are now reaching the marketing stage in Europe and/or in the US. However, only a subset of patients are eligible for those therapies. Many questions remain, such as the duration of the therapeutic effect, the burden of high doses of vectors, and the immunogenicity of viral capsids and therapeutic proteins, in the context of a disease-related inflammatory background. Evaluations of these treatments by the different biotech, pharma or non-for-profit sponsors also come up against the great clinical heterogeneity of patients. This review summarizes the significant progress made over the past three decades to optimize both the efficacy and safety of DMD gene therapies, as well as the remaining challenges, short-term prospects, and future directions such as more targeted vectors and combination therapies.
Keywords
INTRODUCTION: NUMEROUS HURDLES TO OVERCOME
Duchenne muscular dystrophy (DMD) is certainly one of the most complex diseases to address with gene therapy. It piles up a number of barriers and difficulties that decades of research have tried to overcome gradually. It is a rare disease. Its prevalence is currently around 1/5,000 boys worldwide[1]. There are rare cases of female patients or complications for female carriers of recessive mutations present on the X chromosome (OMIM#300377, Xp21.2-p21.1). The relative rarity of the disease hampers large-scale clinical trials.
DMD (MIM #310200), the largest human gene, spans 2.24 million base pairs. Mutations in this gene are the primary cause of the disease. It contains multiple (seven) gene promoters, which drive the expression of different mRNAs and protein isoforms (ranging from 40 kDa to the full-length 427 kDa) in a tissue-specific and time-dependent manner[2]. Clinical manifestations of DMD include progressive muscle degeneration and waisting, typically becoming apparent between the ages of 2 and 5 years. Loss of ambulation often occurs during the early teenage years, followed by the need for respiratory assistance in the early 20s and premature death in the subsequent decade, due to respiratory and cardiac complications[3]. Clinical consequences and pace of disease course may vary depending on the mutation site and the genomic (such as in-frame versus out-of-frame) alterations, which complicate the constitution of clinically homogeneous trial cohorts. Other confounding factors in the assessment of the progression of muscle pathology, with or without treatment, need to be taken into account, such as the initial motor improvement seen in young patients due to age growth and development, which confound statistical interpretations. The first
The disease is multisystemic, affecting various tissues where the protein or its isoforms are expressed: skeletal muscles, vascular and visceral smooth muscle, the heart, endocrine tissues, and the retina, as well as central and peripheral neurons. Therefore, it is essential to ensure the distribution of vectors and their expression throughout the body, even if the priority should be given to the life-threatening skeletal muscles including respiratory muscles and the heart, which together represent 40% to 50% of the body’s mass[6].
Dystrophin is a structural, cytoskeletal membrane protein that plays a critical role as a mechanical link between the extracellular matrix and contractile proteins, providing protection against force-related fiber damage. To achieve this protective function, it is essential not only to target a large number of cells but also to ensure robust and widespread expression of dystrophin. The expression must be sufficient to protect the entire length of muscle fibers from membrane disruptions caused by the mechanical stress of contractions[7].
DMD is a chronic disease, which necessitates that molecular therapies remain effective throughout the patient’s lifetime. This can be achieved either through a single administration or repeated administration of the therapeutic vector, which, if viral, induces an immune response akin to a vaccine, preventing secondary injections and even first-line treatment in seropositive patients. DMD is also a slowly progressive disease, posing challenges for clinical trial design. Trails often require long follow-up periods and highly sensitive clinical endpoints to achieve statistical significance. Consequently, such trials are typically limited to patients with sufficient motor abilities. Additionally, DMD is a developmental disorder with potentially deleterious consequences from the fetal period, which we do not know if post-natal treatment can reverse[8].
Skeletal muscles are regenerative tissues, and DMD is characterized by a succession of necrosis and regeneration cycles, which raises questions about the persistence or the dilution of the transduced vectors over time. The condition involves dystrophic changes, including histological rearrangements and the invasion of fibrotic and fatty tissues (by transdifferentiation of muscle cells, activation of fibro-adipogenic progenitors, or chemoattraction of non-muscular cells)[9], which all create physical and physiological barriers that impede the distribution and efficacy of vector particles. To maximize therapeutic outcomes, intervention must be timed carefully-early enough to prevent irreversible remodeling, yet not so early as to encounter the dilutive effects linked to both regenerative cycles and the rapid muscle growth of children, where muscle mass can increase up to 20-fold after birth. Additionally, the transduction efficiency may be influenced by patient age, as vector biodistribution in skeletal muscles seems to be more favorable at earlier ages[10].
DMD is a chronic inflammatory disease, which is an important limiting factor. Mononucleated cell infiltrates (activated cells that can phagocytose foreign particles) can hinder the ability of vector particles to reach muscle fibers. These infiltrates also pose a risk for inflammatory/immunological overreactions to the administration of vector particles, which are mainly viral. The risk of a robust innate immune response is increased due to the high doses to be administered, not only against the vector but also against previously unrecognized dystrophin epitopes. These immune responses may vary across patients[11]. In particular, patients with deletions in the most immunogenic areas of dystrophin, who are “naïve” to this protein, would be at risk of immunological rejection[12]. This concern is amplified by the fact that dystrophin is expressed throughout the body, especially in vital tissues such as the respiratory muscles, heart, and blood vessels. While a humoral response may not be problematic - since dystrophin is an intracellular protein and thus inaccessible to antibodies - a cellular immune response must be avoided at all costs.
GENE THERAPIES OF DMD
Gene therapy for DMD is complicated by the complex genotype-phenotype relationships associated with the disease. Thousands of different mutations in DMD have been found in DMD patients, including those with Becker muscular dystrophy (BMD), a milder form of the disease. The majority of these mutations (60%-70%) are deletions, 5%-15% are duplications (20% in BMD), and 20% are point mutations (10% in BMD). Deletions and duplications predominantly occur in hotspot regions between exons 45-55 and 3-9, affecting 47% and 7% of patients, respectively[2,13,14]. While some trends in genotype-phenotype correlations have been observed, other factors - such as genetic modifiers, inflammation, and fibrosis invasion - may also influence disease progression, making therapy evaluation particularly challenging. The introduction of molecular therapies would further add to this variability[15].
Several therapeutic technologies have been approved or are under development to target specific dystrophin mutations at the DNA or (pre)-messenger RNA levels. A first targeted therapy conditionally approved by the European Medical Agency (EMA) for DMD was Ataluren, based on pharmacological readthrough of premature stop codons during protein translation. Although it has not yet been approved by the US Food and Drug Administration (FDA), the EMA recommended in June 2024 that the conditional authorization granted to Ataluren in 2014 not be renewed due to insufficient evidence supporting the medicine’s effectiveness[16].
RNA-based therapies
At the pre-messenger RNA level, the conversion of out-of-frame (leading to DMD) to in-frame deletions (associated with milder forms) can be achieved by exon skipping (with oligonucleotides or U7snRNPs expressing an antisense sequence) or by exon deletions (with genome editing) leading to partially functional BMD-like truncated dystrophins. This treatment strategy is mutation-dependent and each individual
Approved RNA-based therapies
Several antisense oligonucleotide (ASO) chemistries have been developed and approved by the FDA and by the Japanese authority (vitolarsen). The current marketed products target mutations amenable to skipping exons 51 (eteplirsen), 45 (casimersen), and 53 (golodirsen and vitolarsen). However, these therapies have not yet been approved by the EMA. ASOs are small modified RNA sequences (20-30 nucleotides) that can be systemically delivered and specifically bind to a target exon during pre-mRNA splicing, preventing the inclusion of the exon into mRNA. Exon skipping can address certain deletions, duplications, and point mutations. However, due to the turnover of both ASO and the transcript and protein, repeated treatments are required[14,18]. More importantly, the ability of these therapies to produce truncated dystrophin remains very limited, with levels typically ranging from 0.4% to 6% of normal dystrophin expression, which are not sufficient for the biological protection of muscle fibers (estimated at 20% to 40%). These numbers are based on numerous studies conducted in both animal models and DMD and BMD patients[20,21]. However, the reliability of these figures is not guaranteed, as they may be influenced by factors such as the functionality of the dystrophin produced, individual variations, and species-specific specificities.
Future directions
Efforts are increasingly focused on developing more efficient chemistries that enhance biodistribution, extend pharmacokinetics (to reduce injection frequency), and target additional exons to expand the pool of eligible patients[18]. The first ASO exon-skipping drugs were limited by low myocardial efficacy and suboptimal pharmacokinetics. To address these limitations, a new chemical approach based on
Duplicated exons, including exon 2, one of the most commonly duplicated exons, are also eligible for exon skipping. Skipping of duplicated exons potentially restores a full-length (and thus fully functional) dystrophin. This approach is currently being investigated using an adeno-associated virus (AAV) vector carrying four copies of a modified U7 small nuclear RNA that contains antisense sequences targeting the splice donor (2 copies) and splice acceptor (2 copies) of the DMD exon 2[24] (NCT04240314).
However, the recruitment of patients with specific genetic subtypes within this already rare disease presents a bottleneck for drug development, which is further complicated by the individual variability in clinical scores over time. Additionally, to optimize statistical power and avoid bias, trial designs should prioritize matching comparative groups based on reliable, data-driven prognostic factors. Recent studies suggest that trial designs should avoid overemphasizing balance in genotype classification[25]. This requires, for instance, to include a natural history/baseline study to monitor clinical and biological evolution for at least 6 months before enrolling DMD patients. Part of the genotype effect may already be captured through baseline functional status, reducing the confounding influence of genotype. This is crucial for any type of therapeutic approach to DMD.
DNA-based therapies
Genome editing
Recently, genome editing has been proposed to restore the reading frame at the DNA level. CRISPR/Cas gene editing may restore full-length dystrophin expression after a single treatment in dystrophic rodent and dog models, although additional work is required to demonstrate the feasibility and safety of this approach in humans. It has several drawbacks such as: (I) very low efficiency in deleting multiple exons, (II) lack of efficiency in delivering the CRISPR-Cas system in vivo, (III) possible need for repeated treatments, and (IV) off-target related safety concerns[26]. In a first-in-man trial, a 27-year-old patient with DMD was treated with a dose of 1014 viral genomes (vg)/kilogram(kg) of body weight of an AAV of serotype 9 containing dSaCas9 (Staphylococcus aureus Cas9 in which the Cas9 nuclease activity has been inactivated). Eight days after infusion, the patient sadly died after an initial mild cardiac dysfunction and pericardial effusion, followed by acute respiratory distress syndrome and cardiac arrest. Given the timelines, genome editing is probably not in question as it did not have enough time to take place. A postmortem examination showed severe diffuse alveolar damage, probably elicited by an innate immune reaction to the high-dose AAV. Transgene early expression was minimal, and there was no evidence of AAV serotype 9 antibodies or effector T-cell reactivity in the organs[27].
Replacement gene therapy
Since DMD is a monogenic disease, gene replacement therapy is one of the most logical and promising treatment options. The first gene therapy clinical trial was completed in 2004. It showed that local intramuscular administration of a low dose (600 µg) full-length dystrophin plasmid in 9 Duchenne and Becker patients allowed only very low and local (along the needle track) expression of dystrophin[28]. Intravascular delivery in isolated limbs (referred to as hydrodynamic limb vein (HLV)-HLV- injection) of the plasmid showed a much higher muscle transfection efficiency (up to 40% of transfected limb muscles) in primate, and in mouse (mdx) and dog (Golden Retriever Muscular Dystrophy) DMD models[29]. Safety of the locoregional HLV delivery of increasing volumes of saline buffer was asserted in a series [limb-girdle muscular dystrophy 2A, Emery-Dreifuss muscular dystrophy, autosomal recessive autosomal dominant Limb Girdle Muscular Dystrophy (LGMD), LGMD, and BMD] of volunteers in lower[30] and upper[31] limbs. However, certain limits were imposed on the volume administered (not exceeding 20%-40% of the limb volume to be administered) due to the internal pressure within the muscle tissues and the theoretical risk of compartment syndrome in already weakened muscles. This limitation on the injected volume posed a barrier to the use of plasmids in DMD.
Viral vectors may transduce skeletal muscle much more efficiently and more broadly. A few of them can accommodate the full-length, 14 kb dystrophin coding sequence together with the associated promoter and regulatory sequences, but they are either ineffective (lentiviruses) or too inflammatory (e.g., poxvirus) to be considered[32,33]. As of today, AAVs outperform other vectors in delivering genes to skeletal muscles and the heart. Moreover, they can infect both dividing and nondividing cells and enable relatively long-lasting and stable gene expression. However, the dystrophin cDNA exceeds the capacity of AAVs (~5 kb).
Microdystrophin gene therapy
The initial discovery that truncated dystrophins retain functionality[34] provided the rationale for using minimized versions of dystrophin compatible with the capacity of AAVs. These highly modified, smaller molecular weight versions of dystrophin, known as microdystrophins (~150 kDa compared to normal
AAVs come in the form of various serotypes that differ in tissue tropism and transduction efficiency that may also be species-dependent[36]. AAV8, AAV9, AAVrh74 (highly similar to AAV8), and Myo-AAV are currently used in clinical trials for DMD. The sarcolemmal dystrophin is organized into highly focal and nonmobile nuclear domains of ~80 µm in size[37], although we cannot exclude the existence of a diffusible cytoplasmic fraction that could be below the level of detection, as well as the contribution of mobile nuclei. Therefore, any AAV gene therapy should transduce as many myonuclei as possible to avoid an ineffective mosaic microdystrophin expression.
As of June 2024, the FDA has approved one product, Elevidys (AAVRh74-hMD1), developed by Sarepta/Roche, for patients aged 4 years and older[38]. The EMA is currently reviewing the application for patients aged 4 to 7 years only. Long-term and extension studies are ongoing (NCT05881408, NCT04626674, NCT05096221, NCT06128564, NCT0596735). Another product developed by Pfizer was recently dropped after missing the primary endpoint in a phase 3 trial (NCT05689164, NCT02907619, NCT05429372). The death of two patients during this study certainly influenced the sponsor’s decision. This product uses an AAV9 vector carrying a different version of microdystrophin[12]. Based on public releases and communications at congresses, we know that Solid Bioscience has also decided to stop the development of its AAV9-microdystrophin (the third version of truncated dystrophin) and switched to a second-generation vector, SGT-003 (NCT06138639) based on Myo-AAV vector with expected reduced immunogenicity and improved efficiency. Additionally, two other sponsors are pursuing the clinical development of two
The degree of functionality and therapeutic impact of microdystrophins in humans remains unclear. The retained portions of the protein and the level of expression may influence its function. Chamberlain et al.[39] and Boehler et al.[40] provide a detailed discussion of critical microdystrophin elements. Additionally, the quantity of microdystrophin expressed may contribute to some aspects of clinical benefit[39]. All sponsors are using different tissue-specific promoters (MHCK7, MHC, K8, spc5.12, and spc2.12)[12]. Therapeutic doses generally exceed 1014 vg/kg except for Genethon (3 × 1013 vg/kg). However, uniform and head-to-head titrations should be performed to accurately assess the differences in viral loads injected. Large-scale production methods also vary. Some use plasmid-transfected HEK293 cells of different strains, while others (such as Solid Biosciences) use an Herpes Simplex (HSV)-based process. Purification methods may also differ[41]. Overall, these variations may result in differences in transduction efficiency, functional benefit, and safety.
AAVs transduce differentiated fibers but not satellite cells as initially reported. Kwon et al., however, reported that satellite cells can be transduced with AAV vectors and can undergo gene editing to restore the dystrophin reading frame in a sensitive Cre/lox-based dual-reporter mdx mouse model. Interestingly, higher AAV transduction of satellite cells was seen in mdx mice compared to Wild Type (WT) mice, supposedly because of the constant need for regeneration and a higher number of activated satellite cells in mdx mice[42].
Safety and durability of AAV-based gene therapy
AAV integration into the host genome
AAV are thought to rarely integrate into the host genome and it is possible that, with successive cycles of degeneration/regeneration (which may occur naturally throughout life), the therapeutic transgene will fade with time due to some dilution effect. Whether this really happens and how long it takes remain unknown, at least in humans. To avoid the risk of gradual loss of episomal transgenes and microdystrophin expression, transgene expression must exceed the threshold (yet unclear) necessary. This has been reported in dystrophic animal models[43,44]. Additionally, the “dilution” effect associated with muscle growth in very young patients suggests that repeated administration of any DMD gene therapy will likely be needed. However, the required interval between treatments and how to circumvent the presence of circulating AAV neutralizing antibodies and the potential T cell response induced by the primary treatment remain to be determined. Nonetheless, studies in Golden Retriever Muscular Dystrophy (GRMD) dog models have shown that long-term (lifelong, > 10 years) clinical benefits[45,46] can still be expected.
Anti-AAV immunity
As seen in the general population[47], many DMD boys may have been exposed to AAVs, potentially resulting in pre-existing immunity to the AAV serotype used for gene therapy[48,49]. Consequently, these patients are currently excluded from receiving gene therapy. To address this limitation, immunosuppressive strategies are being developed[50,51]. For instance, prior administration of IdeS (Imlifidase), a bacterial protease that cleaves human antibodies[52] (NCT06241950), is being investigated in DMD. Systemic AAV gene delivery, with high doses of ~1013 to 1014 vector genomes/kg, has been shown to elicit the activation of the innate immune response to the vector in large animal models and humans, including neuromuscular patients[41]. An adaptive T lymphocyte cell (CTL)-mediated immune response following AAV transduction could lead to the clearance of transduced cells. However, this response is not expected to be detrimental, as healthy skeletal muscle cells exhibit low MHC-I expression, which reduces the T-lymphocyte-mediated reaction[53]. In inflammatory contexts, such as in DMD patients, MHC-I antigens are overexpressed, and muscles contain activated T cells, which would make DMD muscle tissue more susceptible to T-cell immune responses[54]. Additionally, given the high viral load used, a dose-dependent innate immune response eventually initiates adaptive immune responses against both the viral capsid and the transgene. Strategies like CpG depletion from rAAV vector constructs[55] and enhancing vector production by artificially increasing CpG methylation could partially alleviate this anti-AAV innate immune response. Empty capsids may also be used as decoys for anti-AAV neutralizing antibodies, which means that pure full-capsid batches may not necessarily be more effective than mixed preparations[56]. On the other hand, using different AAV strains may offer the possibility of selecting a suitable vector for patients who are seropositive for a certain strain (and therefore, non-eligible for therapy as they would neutralize the injected vector), provided there is no cross-reactivity.
Anti-transgene immune rejection
Recently, five cases of strikingly similar suspected unexpected serious adverse reactions (SUSARs), including life-threatening myositis, were reported in DMD boys aged 7-9 years, who were recruited into three separate microdystrophin gene therapy trials (NCT04281485, NCT04626674,
Level of microdystrophin expression
It is clear that a major clinical goal is to achieve sufficient microdystrophin expression throughout the skeletal and cardiac muscles. However, very little is known about the impact of microdystrophin overexpression. Cardiac toxicity has been reported in transgenic mice with 100-fold overexpression of microdystrophin[59]. Recently, Hart et al.[60] reported the accelerated onset of dilated cardiomyopathy, heart failure, and death in dystrophic mice following AAV-induced overexpression of two of several microdystrophin variants. Further studies are needed to better characterize the potential cardiotoxicity elicited by overexpressed microdystrophin in the heart.
While microdystrophin gene therapy may prove to be a game-changer, it is not expected to fully restore function in skeletal and cardiac muscles. The therapy is theoretically limited to shifting the phenotype from DMD to a mild BMD phenotype, even with high levels of expression. This is supported by current trials, where blood creatine kinase levels - an indicator of skeletal and cardiac muscle damage - seem to drop to BMD levels, but not to those of healthy controls[57].
Alternative transgenes
Full-length or quasi-dystrophin
Researchers are developing dystrophin versions that are closer to the full-length protein, using combinations of AAV vectors and leveraging the ability of AAV genomes to undergo intermolecular concatemerization. A few studies have reported the use of dual AAV vectors for therapeutic correction in DMD, and even a triple-vector system has been used to reconstitute the full dystrophin coding sequence through a trans-splicing system, though with very low efficiency[61,62]. Full-length dystrophin can also be generated using a triple AAV combination based on split inteins - small polypeptides that self-assemble and undergo a protein trans-splicing reaction. This triple-vector combination improved muscle histopathology and function in a mouse model of DMD[63]. However, the translational potential of dual or triple AAV vectors for human patients needs further investigation. For instance, homologous recombination efficiency requires further optimization to reduce the total load of AAV vectors. Additionally, the formation of aberrant products resulting from AAV Inverted Terminal Repeats (ITR) concatemerization requires in-depth evaluation. Vectors that can efficiently transduce satellite cells would be particularly useful for promoting muscle regeneration. Lastly, the quasi-dystrophin produced by this approach may bear a higher risk of inducing an anti-transgene immune response, which must be prevented.
Non-dystrophin genes
To bypass any anti-transgene immune response, genes that are constitutively expressed are being investigated. Utrophin, a functional paralog of dystrophin that is naturally expressed in muscles, has been proposed as an immunologically safer therapeutic gene alternative. Utrophin is a ubiquitous protein, expressed at the sarcolemma of embryonic muscles and progressively replaced by dystrophin at the muscle membrane after birth. In adult skeletal muscle, utrophin is confined to the neuromuscular and myotendinous junctions, as well as blood vessels, and is only expressed at the sarcolemma of regenerated myofibers. Utrophin is highly homologous to dystrophin and can recruit most of the components of the dystrophin-associated protein complex[64]. Despite efforts, pharmaceutical stimulation of utrophin to compensate for the absence of dystrophin has not yet been successful. Therefore, utrophin gene therapy remains a promising alternative. Given the size of its coding sequence, truncated versions of micro-utrophin are being designed, with consideration given to the differential binding capacity and suitable folding of utrophin and dystrophin[65].
Another potential candidate is Cytotoxic T cell GalNAc transferase (GALGT2), an enzyme normally distributed, like utrophin, at the neuromuscular junctions of myofibers postnatally. GALGT2 glycosylates the dystrophin-associated glycoprotein a-dystroglycan. The delivery ofrAAVrh74-GALGT2 has been shown to successfully treat DMD mice[66] and, to a lesser extent, DMD dogs[67]. Encouraging functional data were obtained in a phase 1/2 clinical trial assessing the safety and efficacy of GALGT2 gene therapy (NCT03333590)[68]. However, the clinical development of this program was discontinued by the sponsor[69].
AAV-microdystrophin, utrophin, or GALGT2 gene therapy would be potentially applicable to all DMD patients regardless of their mutation. Active clinical development programs and approved gene-based drugs are shown in Figure 1.
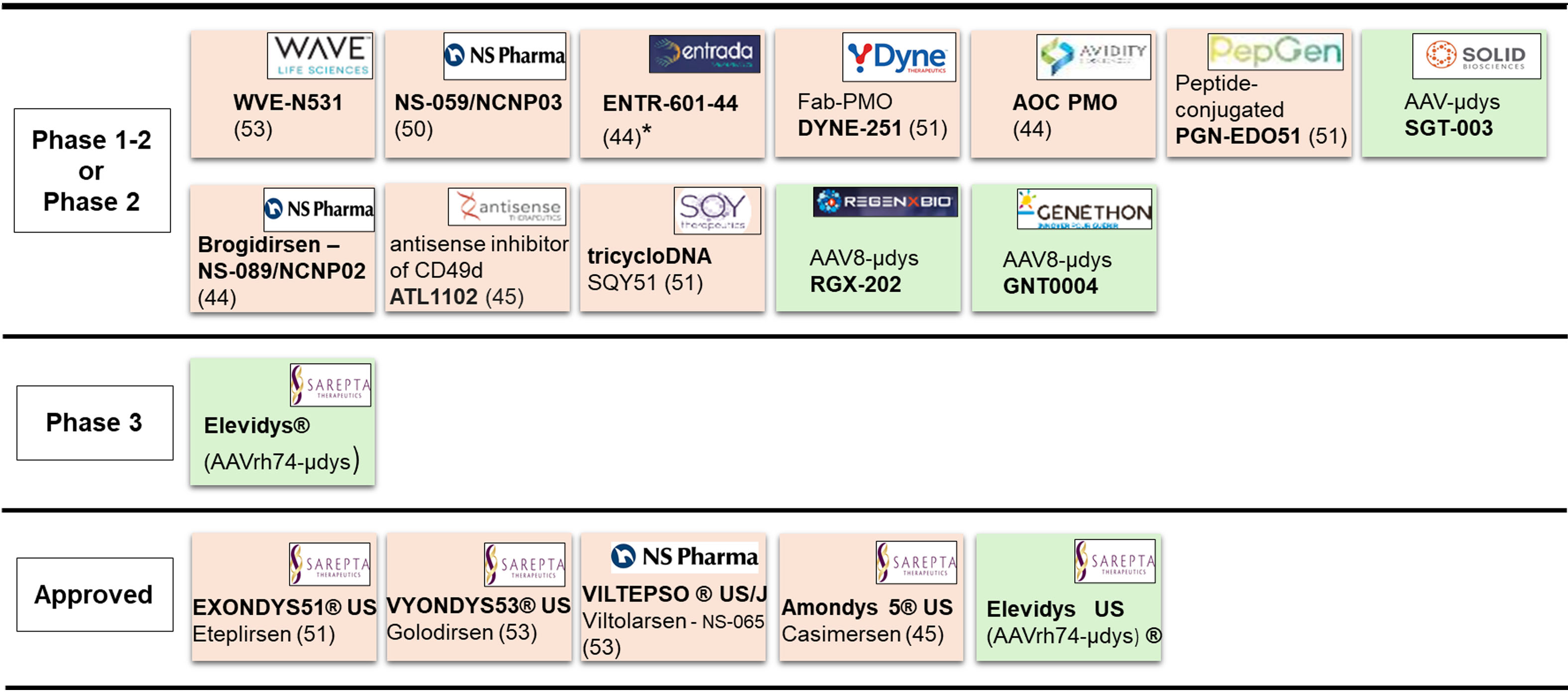
Figure 1. Current pipeline of gene-based therapies. Positioning of the antisense (beige) or gene-replacement (green) therapies, according to the stage of development (left column). Bold: drug name including commercial name (®); US: approved in USA; J: approved in Japan; Logos: name of the sponsor; Number in parentheses: targeted exon; *: clinical hold by FDA; µdys: microdystrophin; Fab-PMO: antibody-conjugated Phosphorodiamidate Morpholino Oligomers. Two other studies carried out in China (NCT06114056 relative to an AAV-microdystrophin, and NCT06594094 relative to a CRISPR-gene editing approach) are not indicated here. PMO: Phosphorodiamidate morpholino oligomer; AAV: Adeno-associated virus; FDA: Food and drug administration.
A range of challenges remain in fully optimizing AAV dosage, immune control and large-scale manufacturing. A more potent expression cassette could reduce the required viral dose to achieve the therapeutic effect. However, according to the nuclear domain theory, stronger expression cassettes with lower vector amounts would not align with the need to transduce a maximum of nuclei along the muscle fibers. Natural variant-based AAVs lack specificity and often accumulate in the liver, with the concomitant risks of hepatotoxicity and general toxicity due to high dosage[70]. One potential solution is lowering the dose by enhancing vector specificity through capsid modification. High-throughput screened or computationally designed AAV capsids that more specifically target skeletal muscle to lower treatment doses are being developed[71]. A recent variant, LICA1, demonstrates comparable muscle transduction to other myotropic AAVs, with significantly reduced liver targeting and, not to underestimate, large-scale production yield capacity[72], which could help reduce the heavy burden of manufacturing costs. Nevertheless, while myotropic AAVs represent attractive second-generation vectors, they still require careful safety assessment.
CONCLUSION
The perfect molecular therapy, administered in very low doses and targeting motor function, circulation, and digestive tracks, as well as cognitive skills, does not exist yet. Nevertheless, emerging combination therapies show promise in addressing the specific needs of each organ. The remaining hurdles (immunological issues, persistence and optimization of therapeutic effects for complete disease remission) are being investigated separately and possible solutions are under consideration. Another key obstacle remains: the development of an economically viable model that guarantees access to these expensive combination biological treatments for all patients[73], including non-ambulant individuals with DMD, BMD, rare female DMD patients, and affected female carriers. Part of the answer relies on scientific and technological progress.
DECLARATIONS
Acknowledgments
The author thanks the Genethon team, AFM-Telethon, and the many external collaborators, including researchers, clinicians, and non-clinicians.
Authors’ contributions
The author contributed solely to the article.
Availability of data and materials
Not applicable.
Conflicts of interest
Genethon is a non-profit institute established by AFM-Telethon, a patient organization.
Financial support and sponsorship
Not applicable.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Salari N, Fatahi B, Valipour E, et al. Global prevalence of Duchenne and Becker muscular dystrophy: a systematic review and meta-analysis. J Orthop Surg Res. 2022;17:96.
2. Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021;7:13.
4. Pane M, Mazzone ES, Sormani MP, et al. 6 Minute walk test in Duchenne MD patients with different mutations: 12 month changes. PLoS One. 2014;9:e83400.
5. McDonald CM, Henricson EK, Abresch RT, et al. PTC124-GD-007-DMD Study Group. The 6-minute walk test and other clinical endpoints in duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. 2013;48:357-68.
6. Topaloglu H. Duchenne muscular dystophy: a short review and treatment update. Iran J Child Neurol. 2021;15:9-15.
7. Elangkovan N, Dickson G. Gene therapy for Duchenne muscular dystrophy. J Neuromuscul Dis. 2021;8:S303-16.
8. Mozin E, Massouridès E, Mournetas V, et al. Dystrophin deficiency impairs cell junction formation during embryonic myogenesis. bioRxiv. 2024; doi: 10.1101/2023.12.05.569919.
9. Sohn J, Lu A, Tang Y, Wang B, Huard J. Activation of non-myogenic mesenchymal stem cells during the disease progression in dystrophic dystrophin/utrophin knockout mice. Hum Mol Genet. 2015;24:3814-29.
10. Bostick B, Ghosh A, Yue Y, Long C, Duan D. Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Ther. 2007;14:1605-9.
11. Mendell JR, Campbell K, Rodino-Klapac L, et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363:1429-37.
12. Bönnemann CG, Belluscio BA, Braun S, Morris C, Singh T, Muntoni F. Dystrophin immunity after gene therapy for Duchenne’s muscular dystrophy. N Engl J Med. 2023;388:2294-6.
13. Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395-402.
14. Sun C, Shen L, Zhang Z, Xie X. Therapeutic strategies for Duchenne muscular dystrophy: an update. Genes. 2020;11:837.
15. Duan D, Goemans N, Takeda S. et al. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021;7:13.
16. EMA recommends non-renewal of authorisation of Duchenne muscular dystrophy medicine translarna. Available from: https://www.ema.europa.eu/en/news/ema-recommends-non-renewal-authorisation-duchenne-muscular-dystrophy-medicine-translarna-0 [Last accessed on 24 Jan 2025].
17. Brun C, Suter D, Pauli C, et al. U7 snRNAs induce correction of mutated dystrophin pre-mRNA by exon skipping. Cell Mol Life Sci. 2003;60:557-66.
18. Takeda S, Clemens PR, Hoffman EP. Exon-skipping in Duchenne muscular dystrophy. J Neuromuscul Dis. 2021;8:S343-58.
19. Leckie J, Zia A, Yokota T. An updated analysis of exon-skipping applicability for Duchenne muscular dystrophy using the UMD-DMD database. Genes. 2024;15:1489.
20. Neri M, Torelli S, Brown S, et al. Dystrophin levels as low as 30% are sufficient to avoid muscular dystrophy in the human. Neuromuscul Disord. 2007;17:913-8.
21. Wells DJ. What is the level of dystrophin expression required for effective therapy of Duchenne muscular dystrophy? J Muscle Res Cell Motil. 2019;40:141-50.
22. Relizani K, Echevarría L, Zarrouki F, et al. Palmitic acid conjugation enhances potency of tricyclo-DNA splice switching oligonucleotides. Nucleic Acids Res. 2022;50:17-34.
23. Cochran M, Marks I, Albin T, et al. Structure-activity relationship of antibody-oligonucleotide conjugates: evaluating bioconjugation strategies for antibody-phosphorodiamidate morpholino oligomer conjugates for drug development. J Med Chem. 2024;67:14868-84.
24. Gushchina LV, Bradley AJ, Vetter TA, et al. Persistence of exon 2 skipping and dystrophin expression at 18 months after U7snRNA-mediated therapy in the Dup2 mouse model. Mol Ther Methods Clin Dev. 2023;31:101144.
25. Muntoni F, Signorovitch J, Sajeev G, et al. Association Française Contre Les Myopathies; on behalf of Universitaire Ziekenhuizen Leuven Group; PRO-DMD-01; The UK NorthStar Clinical Network; CCHMC; and The DMD Italian Group. DMD genotypes and motor function in Duchenne muscular dystrophy: a multi-institution meta-analysis with implications for clinical trials. Neurology. 2023;100:e1540-54.
26. Chemello F, Olson EN, Bassel-Duby R. CRISPR-editing therapy for Duchenne muscular dystrophy. Hum Gene Ther. 2023;34:379-87.
27. Lek A, Wong B, Keeler A, et al. Death after high-dose rAAV9 gene therapy in a patient with Duchenne’s muscular dystrophy. N Engl J Med. 2023;389:1203-10.
28. Romero NB, Braun S, Benveniste O, et al. Phase I study of dystrophin plasmid-based gene therapy in Duchenne/Becker muscular dystrophy. Hum Gene Ther. 2004;15:1065-76.
29. Wooddell C. Dose response in rodents and nonhuman primates after hydrodynamic limb vein delivery of naked plasmid DNA. Hum Gene Ther. 2011;22:889-903.
30. Fan Z, Kocis K, Valley R, et al. Safety and feasibility of high-pressure transvenous limb perfusion with 0.9% saline in human muscular dystrophy. Mol Ther. 2012;20:456-61.
31. Fan Z, Kocis K, Valley R, et al. High-pressure transvenous perfusion of the upper extremity in human muscular dystrophy: a safety study with 0.9% saline. Hum Gene Ther. 2015;26:614-21.
32. Braun S. Gene-based therapies of neuromuscular disorders: an update and the pivotal role of patient organizations in their discovery and implementation. J Gene Med. 2013;15:397-413.
33. Scherman D. Advanced textbook on gene transfer, gene therapy and genetic pharmacology: principles, delivery and pharmacological and biomedical applications of nucleotide-based therapie. 2th ed. Daniel Scherman: Imperial College Press. 2014 pp 17-29.
34. England SB, Nicholson LV, Johnson MA, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343:180-2.
35. Fortunato F, Tonelli L, Farnè M, Selvatici R, Ferlini A. DMD deletions underlining mild dystrophinopathies: literature review highlights phenotype-related mutation clusters and provides insights about genetic mechanisms and prognosis. Front Neurol. 2023;14:1288721.
36. Duan D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol Ther. 2018;26:2337-56.
37. Morin A, Stantzou A, Petrova ON, et al. Dystrophin myonuclear domain restoration governs treatment efficacy in dystrophic muscle. Proc Natl Acad Sci U S A. 2023;120:e2206324120.
38. FDA expands approval of gene therapy for patients with Duchenne muscular dystrophy. Available from: https://www.fda.gov/news-events/press-announcements/fda-expands-approval-gene-therapy-patients-duchenne-muscular-dystrophy [Last accessed on 24 Jan 2025].
39. Chamberlain JS, Robb M, Braun S, et al. Microdystrophin expression as a surrogate endpoint for Duchenne muscular dystrophy clinical trials. Hum Gene Ther. 2023;34:404-15.
40. Boehler JF, Brown KJ, Beatka M, et al. Clinical potential of microdystrophin as a surrogate endpoint. Neuromuscul Disord. 2023;33:40-9.
41. Shen W, Liu S, Ou L. rAAV immunogenicity, toxicity, and durability in 255 clinical trials: a meta-analysis. Front Immunol. 2022;13:1001263.
42. Kwon JB, Ettyreddy AR, Vankara A, et al. In vivo gene editing of muscle stem cells with adeno-associated viral vectors in a mouse model of Duchenne muscular dystrophy. Mol Ther Methods Clin Dev. 2020;19:320-9.
43. Vulin A, Barthélémy I, Goyenvalle A, et al. Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol Ther. 2012;20:2120-33.
44. Le Hir M, Goyenvalle A, Peccate C, et al. AAV genome loss from dystrophic mouse muscles during AAV-U7 snRNA-mediated exon-skipping therapy. Mol Ther. 2013;21:1551-8.
45. Dupont JB, Guo J, Renaud-Gabardos E, et al. AAV-mediated gene transfer restores a normal muscle transcriptome in a canine model of X-linked myotubular myopathy. Mol Ther. 2020;28:382-93.
46. Le Guiner C, Fraysse B, Testault I, et al. More than nine year survival of a GRMD dog after injection of AAV-microdystrophin gene therapy. Mol Ther. 2023;31:pp53.
47. Boutin S, Monteilhet V, Veron P, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21:704-12.
48. Verma S, Nwosu SN, Razdan R, et al. Seroprevalence of adeno-associated virus neutralizing antibodies in males with duchenne muscular dystrophy. Hum Gene Ther. 2023;34:430-8.
49. Zygmunt DA, Crowe KE, Flanigan KM, Martin PT. Comparison of serum rAAV serotype-specific antibodies in patients with Duchenne muscular dystrophy, Becker muscular dystrophy, inclusion body myositis, or GNE myopathy. Hum Gene Ther. 2017;28:737-46.
50. Yang TY, Braun M, Lembke W, et al. Immunogenicity assessment of AAV-based gene therapies: an IQ consortium industry white paper. Mol Ther Methods Clin Dev. 2022;26:471-94.
51. Gross DA, Tedesco N, Leborgne C, Ronzitti G. Overcoming the challenges imposed by humoral immunity to AAV vectors to achieve safe and efficient gene transfer in seropositive patients. Front Immunol. 2022;13:857276.
52. Leborgne C, Barbon E, Alexander JM, et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat Med. 2020;26:1096-101.
53. Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122:23-36.
54. McDouall RM, Dunn MJ, Dubowitz V. Expression of class I and class II MHC antigens in neuromuscular diseases. J Neurol Sci. 1989;89:213-26.
55. Faust SM, Bell P, Cutler BJ, et al. CpG-depleted adeno-associated virus vectors evade immune detection. J Clin Invest. 2013;123:2994-3001.
56. VandenDriessche T, Chuah MK. Vector decoys trick the immune response. Sci Transl Med. 2013;5:194fs28.
57. Mendell JR, Sahenk Z, Lehman K. et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 2020;77:1122-31.
58. Lek A, Atas E, Lin B, et al. Meeting report: 2023 nuscular dystrophy association summit on “safety and challenges in gene therapy of neuromuscular diseases”. J Neuromuscul Dis. 2024;11:1139-60.
59. Yue Y, Wasala NB, Bostick B, Duan D. 100-fold but not 50-fold dystrophin overexpression aggravates electrocardiographic defects in the mdx model of Duchenne muscular dystrophy. Mol Ther Methods Clin Dev. 2016;3:16045.
60. Hart CC, Lee YI, Xie J. et al. Potential limitations of microdystrophin gene therapy for Duchenne muscular dystrophy. JCI Insight. 2024;9:e165869.
61. Kodippili K, Hakim CH, Pan X, et al. Dual AAV gene therapy for Duchenne muscular dystrophy with a 7-kb mini-dystrophin gene in the canine model. Hum Gene Ther. 2018; 3:299-311.
62. Albini S, Palmieri L, Dubois A, Bourg N, Lostal W, Richard I. Assessment of therapeutic potential of a dual AAV approach for Duchenne muscular dystrophy. Int J Mol Sci. 2023;24:11421.
63. Zhou Y, Zhang C, Xiao W, Herzog RW, Han R. Systemic delivery of full-length dystrophin in Duchenne muscular dystrophy mice. Nature Com. 2024;15:6141.
64. Tinsley J. M., Davies K.E. Utrophin: a potential replacement for dystrophin? Neuromuscul Disord. 1993;3:537-9.
65. Szwec S, Kaplucha Z, Chamberlain JS, Konieczny P. Dystrophin- and utrophin-based therapeutic approaches for treatment of Duchenne muscular dystrophy: a comparative review. BioDrugs. 2024;38:95-119.
66. Xu R, Jia Y, Zygmunt DA, Martin PT. rAAVrh74.MCK.GALGT2 protects against loss of hemodynamic function in the aging mdx mouse heart. Mol Ther. 2019;27:636-49.
67. Martin PT, Zygmunt DA, Ashbrook A, et al. Short-term treatment of golden retriever muscular dystrophy (GRMD) dogs with rAAVrh74.MHCK7.GALGT2 induces muscle glycosylation and utrophin expression but has no significant effect on muscle strength. PLoS One. 2021;16:e0248721.
68. Flanigan KM, Vetter TA, Simmons TR, et al. A first-in-human phase I/IIa gene transfer clinical trial for Duchenne muscular dystrophy using rAAVrh74.MCK.GALGT2. Mol Ther Methods Clin Dev. 2022;27:47-60.
69. Notice regarding impairment loss for products under development. Available from: https://www.astellas.com/en/news/25731 [Last accessed on 24 Jan 2025].
70. Hordeaux J, Lamontagne RJ, Song C, et al. High-dose systemic adeno-associated virus vector administration causes liver and sinusoidal endothelial cell injury. Mol Ther. 2024;32:952-68.
71. Weinmann J, Weis S, Sippel J, et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat Commun. 2020;11:5432.
72. Hong AV, Seul L, Petat E, et al. An engineered AAV targeting integrin alpha V beta 6 presents improved myotropism across species. Nat Commun. 2024;15:7965.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright















Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].