

Integrating computational materials science and materials informatics for the modeling of phase stability

Abstract
With rapid developments in big data and artificial intelligence technologies, materials informatics has become a new paradigm of materials science and engineering. In this review, the progress of modeling studies of phase stability in alloys is presented, with particular attention given to the development of the paradigm from traditional computational materials science (CMS) to materials informatics. The features of CMS models for phase stability studies are compared with those of data-driven approaches. The advantages of data-driven modeling in the framework of materials informatics are revealed. The approaches for developing interpretable machine learning, which has been mainly integrated with the developed CMS models and material science theories, are also discussed. Finally, the prospects for data-driven materials design based on the stability control of the dominant phases with regards to performance are proposed.
Keywords

Introduction
The mechanical and functional properties of materials are largely dependent on their phase constitutions and microstructures, which are dominated by the design of components and preparation processes[1,2]. The stability of the dominant phases is significant for achieving excellent material properties and maintaining these properties due to the stabilized phase structure. Studies of phase stability and transformation behavior facilitate our understanding of the correlation between phase constitution and materials performance and the development of materials with excellent properties and high-stability dominant phases[3]. Essentially, phase stability is mainly determined by the composition, structural features and energy state of the phase[4].
Phase stability can be investigated by computational methods. Computational materials science (CMS), which is an interdisciplinary subject that traverses both materials science and computer science, is a subject that uses computation and simulation technologies to study the composition, structure and properties of materials[5]. Since the rise of CMS theories and methods in 1980s[6], the development of materials design has been greatly promoted. CMS methods, including first-principles calculations[7], molecular dynamics[8], Monte Carlo simulations[9], phase-field simulations[10], CALPHAD (calculations of phase diagrams)[11,12] and the finite element method[13], have been well developed and play important roles in various materials and at microstructural scales. However, for traditional CMS methods, which are generally used at certain length scale of material microstructures, the computation time may be largely extended with the increase of element types and complexity of phase constitutions and crystal structures in the material.
With the accumulation of experimental and calculation data from research, the development of database technology and the integration of computing and big data techniques, the field of materials informatics has rapidly developed in recent years[14-28]. As proposed by Zhang et al.[14,15], materials informatics integrates techniques, tools and theories drawn from a variety of fields, such as data science, the internet, computer science and engineering, as well as digital technologies applied to materials science and engineering to accelerate materials, products and manufacturing innovations. Materials informatics has now become a new paradigm for materials science research[29-31]. In the framework of Materials Genome Engineering (MGE)[32-34], which seeks to boost high-efficiency materials design, high-throughput calculations and big data technologies continue to be demonstrated for applications in energy materials[35], biomedical materials[36], rare-earth functional materials[37], catalytic materials[38], superalloys[39] and other material systems. A number of MGE technologies have achieved important breakthroughs, for example, in the establishment of models and algorithms[40,41], the development of specific databases[42] and the invention of high-throughput experimental techniques[43,44].
In this review, we provide an overview of modeling studies of phase stability in alloys, including theories, models and methods. The Sm-Co alloy system, as one of the two most famous rare-earth permanent magnetic materials families[45], is taken as an example, with the consideration that this system is rich in various stable and metastable phases under the conditions of binary compositions[46-53]. Efforts are made to introduce the progress in the studies of the phase stability and transformation behavior of Sm-Co-based alloys, in which the development of the research approaches from the traditional CMS methods to strategies based on the materials informatics is particularly demonstrated. Moreover, the advantages and prospects of extending new approaches for studying phase stability to additional research challenges are discussed, including shortening of the research and development period, increasing the modeling accuracy, reducing the costs of experimental trial-and-error processes and accelerating the development of new high-performance materials.
Phases in Sm-Co alloys
In recent decades, significant progress has been made in the field of rare-earth permanent magnetic materials. Sm-Co alloys have irreplaceable advantages in this regard, including high Curie temperatures, good thermal stability and excellent magnetic performance at high temperatures[54]. Thus, Sm-Co alloys are the most promising candidates for applications in critical fields such as aerospace, marine power plants and military apparatus[55]. The Sm-Co system is rich in phases with various crystal structures[56], as shown in Figure 1. There is a series of intermetallic compounds in the Sm-Co system with different stoichiometric ratios, including stable phases such as Sm3Co, Sm9Co4, SmCo2, SmCo3, Sm2Co7, SmCo5 and Sm2Co17, and metastable phases such as Sm5Co19, SmCo7 and SmCo9.8[57-62]. These various phases have different thermodynamic stabilities and the transformation between them certainly influences the magnetic properties of Sm-Co alloys.

Figure 1. Binary phase diagram of Sm-Co system[56].
Increasing attention has been devoted to the research and development of nanoscale rare-earth permanent magnetic materials in recent years[46,63] and these exhibit superior properties compared with their conventional coarse-grained counterparts. A high coercivity can be obtained in a nanostructured alloy because the magnetization reversal is strongly pinned by the grain boundaries that are extremely increased with the decrease of grain size in nanocrystalline materials[46]. In addition, some Sm-Co phases that are metastable in the conventional coarse-grained alloy system can be stabilized in the nanocrystalline system due to the nanoscale effect on the phase stability[64]. Thus, it is very promising to moderate phase stabilities by tailoring the grain size in the Sm-Co system, thereby improving the magnetic performance of the material[49]. In the following sections, progress in the studies of phase stability in Sm-Co alloys is reviewed with regards to modeling approaches from traditional CMS calculations to innovative materials informatics.
CMS calculations of phase stability
Thermodynamic models
Equilibrium phases
A number of investigations have revealed that nanocrystalline materials may have abnormal phase stabilities that result in different phase structures[65-74] compared with their coarse-grained counterparts with the same composition. An earlier thermodynamic calculation reported that for the typical γ-Fe (FCC) ↔ α-Fe (BCC) transformation, if the grain size is sufficiently small, e.g., in nanocrystalline Fe, the γ-Fe phase, which is normally stable at high temperatures, will be stabilized at room temperature[75]. With decreasing grain size, grain boundaries play an increasingly important role in the phase stability of nanocrystalline materials[76]. In a universal model proposed for nanocrystalline metals and single-phased alloys[77], the total Gibbs free energy of the system was calculated as the summation of the energies of the crystalline and interfacial components. Based on a dilated crystal model[78,79], the state of the interfaces in a nanocrystalline system was described in terms of its excess volume and thickness. Similarly, the decrease in the melting and evaporation temperatures with the particle size of the metals was also attributed to the nanoscale effect on the phase transformations. It was considered that the allotropic transformation of Ag nanoparticles is size dependent and the total Gibbs free energy is greatly affected by the surface energy and stress[80]. Therefore, thermodynamics developed for nanoscale materials are required to fill the gap between macroscopic classical thermodynamics and microscopic quantum thermodynamics and thus to interpret abnormal phase transformation phenomena in nanocrystalline materials.
From the view of thermodynamics, a large number of disordered atoms at the grain boundaries significantly influence the entropy, enthalpy and Gibbs free energy of a nanocrystalline material, leading to different thermodynamic functions of the system compared to conventional coarse-grained materials. It is known that the Gibbs-Thomson equation relates the chemical potential to the radius of curvature and energy of the interface[81], which describes the state of a given interface. To derive the thermodynamic functions of the enthalpy, entropy and Gibbs free energy of a nanocrystalline bulk system, a model that considers the effects of grain size and temperature should be developed. In a classic thermodynamic model for nanocrystalline materials[77], an “excess volume”, ΔV, was introduced to characterize the structural features of nanograin boundaries. This volume is directly related to the grain size, d, and can be estimated by[77]:
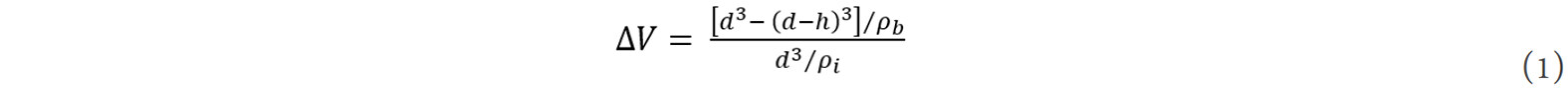
where ρi and ρb are the spatial distribution densities of atoms in the grain interior and at the grain boundary, respectively, d is equivalent to the diameter of a sphere with the same volume of the grain and h is the thickness of the grain boundary.
The ratio between the atomic densities at the grain boundary and in the grain interior can be expressed as[48]:
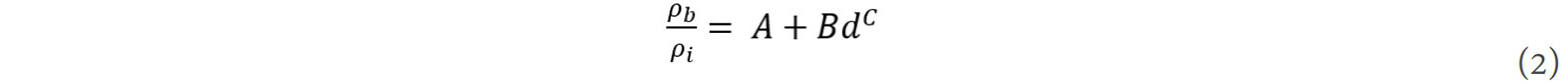
where A, B and C are the fitting coefficients. The unit cell volume at the grain boundary, Vb, can then be expressed as:
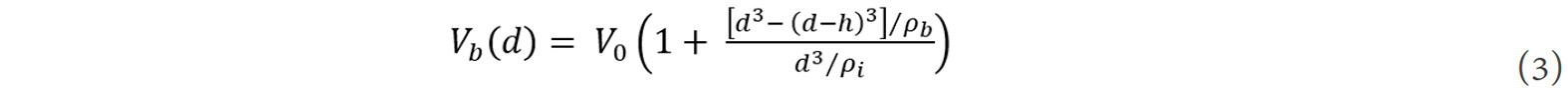
where V0 is the unit cell volume in a perfect crystal. The fundamental thermodynamic functions, i.e., enthalpy, entropy and Gibbs free energy, of the grain boundaries are then given by[48]:
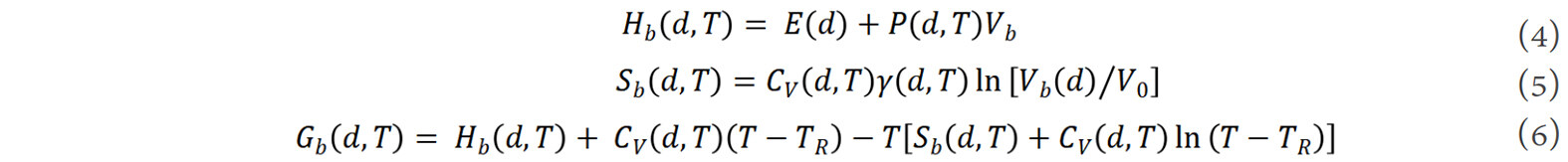
where E is the binding energy of atoms at the grain boundary, P is a negative pressure generated at the grain boundary as a result of the reduction of the grain size[69], CV is the specific heat capacity at constant volume, γ is the Grüneisen parameter of the dilated crystal at the grain boundary[79] and TR is the reference temperature. Thus, the thermodynamic functions of a nanocrystalline alloy can be described as the weighted average of the corresponding functions of the grain boundary and interior components[48]:
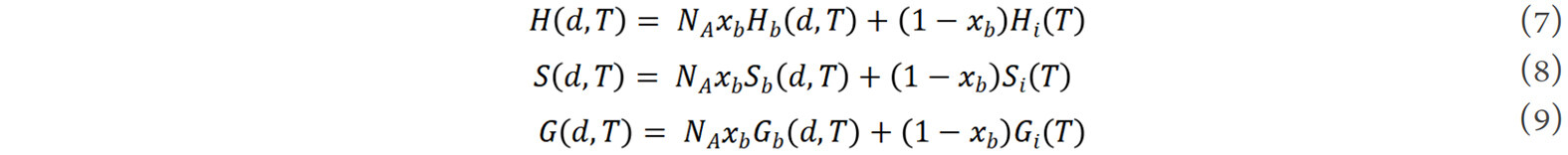
where xb is the volume fraction of grain boundaries, NA is the Avogadro constant, Hi, Si and Gi are the enthalpy, entropy and Gibbs free energy of the grain interior, respectively. From the model, the thermodynamic properties and hence the phase stability and phase transformation characteristics are deterministic functions of grain size and temperature.
By applying the above model, Xu et al.[64,67,71-74] calculated the phase stabilities of a series of nanocrystalline Sm-Co binary alloys and the tendency of phase transformations was then predicted. As an example, in coarse-grained alloys, Sm2Co17 has a stable rhombohedral 2:17R phase at room temperature, while the 2:17H phase is only stable at temperatures higher than 1520 K[74]. However, as predicted by the model calculations shown in Figure 2A and B[74], with decreasing grain size, the Gibbs free energy differences (ΔGH-R) between hexagonal and rhombohedral Sm2Co17 decrease at different temperatures. There exists a critical grain size corresponding to ΔGH-R = 0, implying the critical condition of grain size for the phase stability at a given temperature. In other words, if the grain size is smaller than the critical value, the 2:17H phase can be stabilized at a lower temperature than the phase transformation point of the conventional coarse-grained Sm2Co17 alloy. Moreover, the 2:17H may become the dominant phase at room temperature when the grain size is sufficiently small.

Figure 2. Thermodynamic calculations and experimental verifications of Sm2Co17 alloys with different grain sizes[74]. (A) Calculated Gibbs free energy differences between hexagonal (2:17H) and rhombohedral (2:17R) phases as a function of grain size at different temperatures. (B) Calculated Gibbs free energies of nanocrystalline 2:17H and 2:17R phases as a function of grain size at 1000 K. (C) Transmission electron microscopy images, electron diffraction patterns and indexing of nanocrystalline Sm2Co17 bulk samples in the as-prepared state (C1), annealed at 973 K for 1 h (C2) and annealed at 1073 K for 1 h (C3), respectively.
Experiments were carried out to verify the model calculations in Refs.[48,74]. As shown in Figure 2C, for the prepared bulk alloy with a nominal composition of Sm2Co17 and an average grain size of 15 nm, which was smaller than the model-predicted critical value of 30 nm, the sample had a stable 2:17H phase instead of 2:17R at room temperature, as indicated by transmission electron microscopy[74]. For the sample annealed at 973 K for 1 h, the average grain size was increased to 45 nm and the sample had a mixture of the 2:17H and 2:17R phases at room temperature. When the sample was annealed at 1073 K for 1 h, the average grain size was 80 nm and the alloy had a single 2:17R phase at room temperature. Therefore, the experimental results confirmed the model calculations for the grain size-dependent phase constitution at room temperature in the Sm2Co17 alloy system. This model has been applied to a series of equilibrium (thermodynamically stable in the binary phase diagram) phases in the Sm-Co system, e.g., SmCo5, SmCo2, Sm2Co7, SmCo3 and Sm9Co4 alloys[64,72,73], to evaluate their phase stabilities and transformation behavior at both the micro- and nanoscales.
Metastable phases
Metastable phases in the Sm-Co system, such as SmCo7, Sm5Co19 and SmCo9.8, have been successfully prepared by some non-equilibrium techniques developed in recent years[47,61,62,82,83]. The metastable phase dominated alloys can have special and outstanding functional properties, which may be difficult to obtain in equilibrium phased alloys. However, the metastable phases are generally destabilized at higher temperatures due to their thermodynamically non-equilibrium state. In conventional coarse-grained alloy systems, various doping elements are often used in experiments to stabilize the metastable phases[84-92]. A few theoretical studies have been reported in for modeling the phase stability in metastable phased alloys[93,94]. Unfortunately, the covered alloy systems and microstructural scales by modeling are still limited.
In a developed thermodynamic model[64], it was proposed that the degree of the stability, or the relative stability, could be described by a parameter of phase activity, which had a value from 0 to 1. The activity of a phase, ai, was introduced to characterize the chemical potential difference between a component in the alloy system (μi) and the component at the reference state 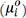 [95]:
[95]:
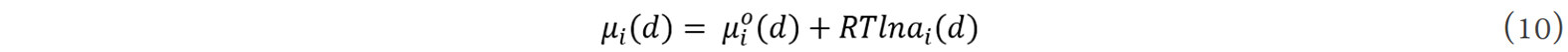
where R is the gas constant. The chemical potential is the partial differential of the Gibbs free energy of the system, 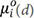 is the chemical potential of a single phase with a grain size of d at 298 K and 1 atm. It can be seen that ai = 1 means the component i is at a thermodynamic equilibrium state in the system and the phase has a highest relative stability, i.e., component i can exist in the form of a stable phase in the system. When other factors are constant, with a change in the grain size, the standard chemical potential of the components in the system also changes. Thus, the stability, destabilization criterion and transition rule of metastable phases can be quantitatively described by the phase activity.
is the chemical potential of a single phase with a grain size of d at 298 K and 1 atm. It can be seen that ai = 1 means the component i is at a thermodynamic equilibrium state in the system and the phase has a highest relative stability, i.e., component i can exist in the form of a stable phase in the system. When other factors are constant, with a change in the grain size, the standard chemical potential of the components in the system also changes. Thus, the stability, destabilization criterion and transition rule of metastable phases can be quantitatively described by the phase activity.
Figure 3 shows the calculated results of the phase activities in the system, taking the typical metastable phase of SmCo7 as an example[95]. The range of grain sizes for the calculation covered from nanocrystalline to a conventional coarse-grained system. In a coarse-grained structure (d > 100 nm), the phase activity of SmCo7 is much lower than a = 1 at room temperature [Figure 3A]. In contrast, the activities of the Sm2Co17 and SmCo5 are a = 1 and the mole fractions of these phases contribute to the total amount of the phases in the system [Figure 3B]. The results indicate that the SmCo7 phase cannot exist stably in the coarse-grained system and at room temperature it decomposes into the Sm2Co17 and SmCo5 phases. The calculations of the metastable phase model based on the concept of phase activity explained the thermodynamic nature of the instability of the SmCo7 phase in the coarse-grained alloys.

Figure 3. Model calculations based on phase activity[95]. (A) Phase activities in the SmCo7 system at room temperature as a function of grain size. (B) Mole fractions of different phases in the SmCo7 system at room temperature.
For a nanocrystalline system (d < 100 nm), the activity of the SmCo7 phase increases rapidly with decreasing grain size. When the grain size is reduced to below 51 nm[95], the phase activity changes from < 1 to 1, indicating that the nano-grained SmCo7 can exist stably as a single phase at grain sizes smaller than 51 nm. Figures 4 and 5 show the experimental results of the phase constitutions and grain structures in the nominal SmCo7 alloys[49,96]. The average grain sizes of the as-prepared nanocrystalline alloy and the sample annealed at 873 K for 0.5 h were 20 and 35 nm, respectively[95], which were both smaller than the predicted critical grain size (51 nm). Thus, the SmCo7 phase was found to exist stably as a single phase in these samples [Figure 4A and B]. After the sample was annealed at 973 K, the average grain size was increased abruptly to 161 nm and it was observed that the SmCo7 phase was partially decomposed into SmCo5 and Sm2Co17, which can be distinguished by the very fine nanoparticles and microtwins in Figure 4C. In particular, Figure 5 shows an individual grain with a size of 166 nm and it has a single phase of 2:17R[96]. This confirms that when the grain size is larger than the critical value, the SmCo7 phase is destabilized and transforms to the equilibrium 2:17R phase. Therefore, with the experimental confirmation, the thermodynamic model based on the phase activity can evaluate the phase stability and transformation tendency of metastable phases.

Figure 4. Experimental investigations of the phase constitutions in SmCo7 samples with different grain sizes[95]: (A) as-prepared state with an average grain size of 20 nm; (B) sample annealed at 873 K for 0.5 h with an average grain size of 35 nm; and (C) sample annealed at 973 K for 0.5 h with an average grain size of 161 nm.

Figure 5. Transmission electron microscopy images and diffraction analysis of a grain with a size of 166 nm[96]: (A) bright-field image; (B) corresponding diffraction pattern and indexing, triangles indicating the 2:17R superstructure reflections; and (C) dark-field image reflecting the twin variants (displayed as red and green), indicating the single 2:17R phase of the whole grain.
First-principles calculations
It is known that doping is an alternative method to stabilize metastable phases in alloys[84-92]. The dopants can change the interactions between the matrix elements, the chemical environment and the system energy and thus influence the phase stability significantly. Considering the different types and contents of the doping elements, as well as the complex interactions between the dopants and the matrix elements, the selection of appropriate doping elements is challenging in materials design. Meanwhile, it is usually difficult to explain the mechanisms of the doping effect on the phase stability only by experimental investigations. In an earlier theoretical study[87], parameters such as formation enthalpy, difference in atomic radius and electronegativity were used to estimate the stability of Sm(Co,M)7 (M is the doping element). However, in the calculations where more empirical or semi-empirical parameters were used, the modeling results were seldom verified by experimental tests.
In recent years, first-principles calculations based on density functional theory were carried out to study the structural stability of multicomponent alloys and the interactions between the electrons of the matrix and doping elements. Compared with thermodynamic models, first-principles calculations can be used to not only evaluate the crystal structure stability of a certain phase, but also to visualize the interactions between different atoms, so as to explain the mechanisms of doping at the atomic scale. In first-principles models, the optimization of the unit cell structure of the crystal is generally first performed and then some specific energies, such as the formation, interface and Gibbs free energies, are calculated. For example, the formation energy of a Sm(Co,M)x phase can be defined as[97,98]:
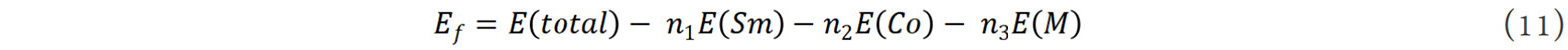
where E(total) is the total energy of the supercell, E(Sm), E(Co) and E(M) are the static energies of Sm, Co and doping element M, respectively, and n1, n2 and n3 are the atomic numbers of the corresponding elements in the supercell.
The formation energies of the binary and Hf-doped SmCo7 phases are shown in Figure 6A, where for the ternary SmCo6.75Hf0.25 phase, different doping sites were considered in the calculations[97]. The results show that when the Hf atoms occupy the Co-2e site, the Hf-doped SmCo7 phase is the most stable. Compared with the binary SmCo7 phase, the decrease in the formation energy of the Hf-doped SmCo7 phase indicates that the doping of Hf may stabilize the metastable SmCo7 phase. In addition to the energy state, temperature also influences the site occupation of the doping elements. The site occupation probability Pi is a function of temperature based on the Maxwell-Boltzmann statistical distribution and can be expressed as[97]:

Figure 6. First-principles calculations for the SmCo7 alloy with Hf doping[97]. (A) Formation energies calculated for SmCo7 and SmCo6.75Hf0.25 with different doping sites. (B) Calculated occupation probability as a function of temperature for the SmCo6.75Hf0.25 phase. (C) Total charge density distributions of SmCo7 (C1) and SmCo6.75Hf0.25 (C2) on (100) plane. (D) Total density of states and partial density of states of SmCo6.75Hf0.25.
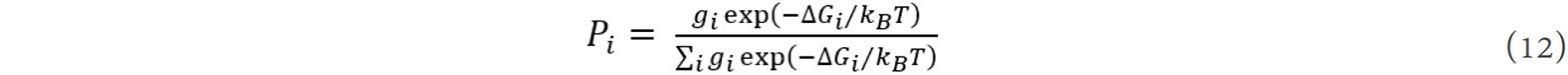
where gi is the multiplicity of configuration i (number of equivalent configurations in the unit cell), ΔGi is the change of the Gibbs free energy and kB is the Boltzmann constant.
The calculations showed that Hf occupation at the 2e site has the highest probability at temperatures below 700 °C [Figure 6B]. When the temperature is higher than 700 °C, the probability of occupations at the 2c and 3g sites increases; however, the value is much smaller than that of 2e, implying that Hf atoms still occupy the 2e site with a high probability. From the total charge density distributions of SmCo7 and SmCo6.75Hf0.25 on a certain crystal plane [Figure 6C], the bond interactions and electronic configurations between different elements can be investigated[97]. It can be seen that the electrons accumulate around Co atoms when the Co-2e atom is partially replaced by a Hf atom, which implies a strong electronic interaction between Co atoms. Comparing Figure 6C1 and C2, the doping of Hf resulted in electron accumulation between Co(2e) atoms and Co(2e)-Sm atoms, thus strengthening the Co(2e)-Co(2e) dumbbell pair atoms and promoting the formation of the Co(2e)-Sm bond. Consequently, the stability of the SmCo7 phase structure can be enhanced by Hf doping.
Due to the spin polarization, the peak of the density of states splits into spin-up and spin-down states, as shown in Figure 6D[97]. The density of states of the electrons at the Fermi energy is set as zero. The ferromagnetic behavior is considered to originate from the asymmetry of the up and down density of states in the upper valence band near the Fermi level[97]. The magnetic moments are mainly contributed to by the 3d electrons of Co atoms and the 4s and 4f electrons of Sm atoms. The doping of Hf leads to an increase in the symmetry of the total density of states [Figure 6D] and thus the total magnetic moments decrease in the Hf-doped alloy.
Experiments confirmed the first-principles calculations, as shown in Figure 7. It was found that the decomposition temperatures of the SmCo7 phase in the SmCo7, SmCo6.85Hf0.15 and SmCo6.75Hf0.25 alloys were 700, 800 and 850 °C, respectively[49,99]. Clearly, the stability of the SmCo7 phase was improved by Hf doping. From modeling and experimental verification, it can be considered that first-principles calculations are effective for evaluating the interactions between the doping elements and the matrix and are thus able to analyze the structural stability of different phases.

Based on first-principles calculations, as shown in Figure 8A, Das et al.[100] found that the formation energy of SmCo5 increased with increasing Fe doping content, while the magnetocrystalline anisotropy energy exhibited the opposite tendency. Moreover, doping with Ni can reduce the formation energy of SmCo5 and the co-doping of Fe and Ni causes the formation of the SmCoNiFe3 phase, which has a high saturation magnetization and stable CaCu5-type crystal structure[101]. In addition, it was found that there exists a negative correlation between the formation energy and the number of 3d electrons [Figure 8B]. Gavrikov et al.[102] performed thermodynamic calculations on the heat of formation of the Sm(Co1-x-yFexNiy)5 phase using homemade software in the framework of Miedema’s model, as shown in Figure 8C and D. The energy states corresponding to 3d ions equiprobably distributing at 2c and 3g sites [Figure 8C] and selectively distributing at lattice sites (Co/Ni at 2c and Fe at 3g, Figure 8D) were compared and the results indicated that the equiprobable distribution of 3d ions is a more accurate model for the calculation of the heat of formation of the co-doped SmCo5 phase.

Figure 8. First-principles calculation results and analysis from Das et al.[100], Söderlind et al.[101], Gavrikov et al.[102]. (A) Calculated magnetocrystalline anisotropy and formation energies of the SmCo5-xFex phase as a function of Fe content. (B) Calculation and experimental results of formation energies as a function of the number of 3d electrons. (C) Concentration diagram of the heat of formation (ΔHf) of Sm(Co1-x-yFexNiy)5 for equiprobably distributed 3d ions at 2c and 3g sites. (D) ΔHf of Sm(Co1-x-yFexNiy)5 for selectively distributed Co/Ni (2c) and Fe (3g) 3d ions. The graded blue regions in (C) and (D) correspond to the negative values of the heat of formation.
Numerical computations
As introduced in the former section, first-principles calculations can be used to evaluate the atomic interactions and hence the effect of doping elements on the structural stability and even material properties. However, it is generally difficult in first-principles calculations to build an accurate model for disordered crystal structures that are often formed due to doping[93]. In this respect, some methods based on interatomic potentials, which are able to deal with millions of atoms, have been developed. A concise inverse method was proposed by Chen[103,104] based on the modified Möbius inverse transformation in number theory. It assumed that the total cohesive energy per atom in a perfect crystal, E(x), could be expressed as the sum of pair potentials, Φ(x), i.e.,[105]:
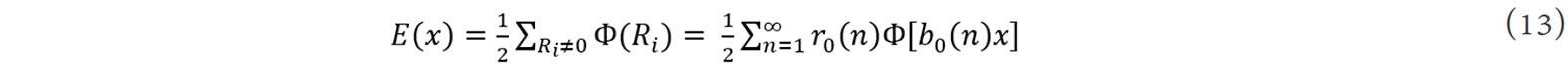
where Ri is the lattice vector of the ith atom, x is the nearest-neighbor interatomic distance, r0(n) is the nth neighbor coordination number and b0(n)x is the nth neighbor distance. By a self-multiplicative process of the element in {b0(n)}, the {b(n)} forms a closed multiplicative semi-group. The general equation for the interatomic pair potential obtained from inversion can then be expressed as[105]:
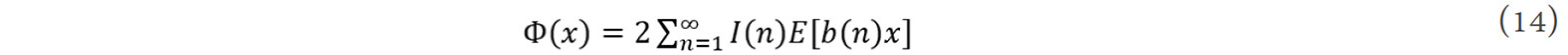
where I(n) is the inversion coefficient, which is uniquely determined by the geometric structure of the crystal. The interatomic pair potentials can then be obtained from the cohesive energy function, E(x).
In order to obtain the necessary interatomic potentials, some virtual structures were designed, e.g., bcc Co was proposed as a B2 or CsCl structure with two simple cubic (SC) sub-lattices, Co1 and Co2. Thus, the cohesive energy of Co-Co was obtained as[106]:
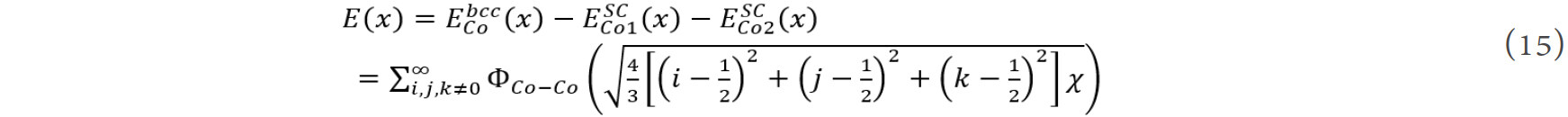
where x is the nearest-neighbor distance in the bcc structure and 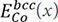 ,
, 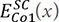 and
and 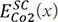 represent the total energies with the bcc and SC structures, which can be calculated by first-principles calculations. Thus, the pair potentials between identical atoms, ΦCo-Co, can be obtained directly using Chen’s lattice inversion technique. In the same way, all other kinds of interatomic potentials can be obtained, as shown in Figure 9A and B[105]. The interatomic pair potential can be fitted by the Morse function[105]:
represent the total energies with the bcc and SC structures, which can be calculated by first-principles calculations. Thus, the pair potentials between identical atoms, ΦCo-Co, can be obtained directly using Chen’s lattice inversion technique. In the same way, all other kinds of interatomic potentials can be obtained, as shown in Figure 9A and B[105]. The interatomic pair potential can be fitted by the Morse function[105]:

Figure 9. Pair potentials for Sm(Co,M)12 as a function of interatomic distance, M = (A) Ti or (B) V. Average energy of SmCo12-xMx with different doping sites as a function of the content of doping element, M = (C) Cr, (D) V, (E) Nb and (F) Ti[105].
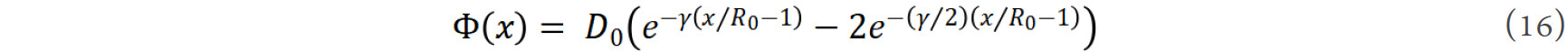
where D0 is the depth of the potential, R0 is the equilibrium distance and γ is a parameter. The pair potential is a function of interatomic distance [Figure 9A and B]. When a small amount of doping element M substitutes Co atoms in the SmCo12 lattice, the M atom is mostly surrounded by the Co atoms. It is the difference between ΦCo-M(r) and ΦCo-Co(r) that determines the energy difference caused by the substitution. If ΦCo-M(r) < ΦCo-Co(r), it is favorable for the energy decrease and the substitution. As the interatomic pair potentials are obtained by a strict lattice inversion of crystal cohesive energy curves, it can be deduced that the pair potentials can properly reproduce the cohesive energy[105].
As shown in Figure 9C-F[105], the preferential occupation of the doping elements Cr, V, Nb and Ti at the 8i site result in the largest energy decrease in SmCo12-based alloys and the ThMn12 structure can be stabilized. This model was also applied successfully to the evaluation of the preferential occupation and stabilization of the doped SmCo7 alloy. The calculations predicted that Ti, Zr and Hf prefer to occupy the 2e site and Ga, Si and Cu prefer to occupy the 3g site[93].
Machine learning of phase stability
As described above, thermodynamic, first-principles and numerical computations are all modeling methods for individual microstructural scales and specific compositions and crystal structures. It is very difficult for these methods to perform high-throughput calculations and predictions for material systems covering a broad range of compositions and phase structures, as well as phase stabilities and transformation characteristics of the systems with various doping elements. Due to the complexity of the cell structure and the diversity of the doping sites, the computation period is usually long. For permanent magnetic materials, to obtain sufficient information of the cell structure of a certain phase in the doped system and the related electronic and magnetic structures, the all-electron method should be used to solve the wavefunction[100], which is extraordinarily time-consuming. However, other methods of first-principles calculations generally have poor convergence when dealing with 4f electrons. As a result, first-principles and thermodynamic calculations of the permanent magnetic materials are limited only to some specific systems and are difficult to carry out for large-scale calculations covering more factors and with a high computational efficiency.
The features of materials informatics enable it to bridge the gap between multiscale models in the study of phase stability. In particular, data-driven models and methods play a significant role in the development of new materials and are attracting increasing attention in materials science and engineering[107-109]. The prerequisite for data-driven materials design is to build high-quality datasets. The sources of the datasets can be divided into two categories, one is from high-throughput computing or experiments and the other is from published research studies. The first type of data is basically dominated by the development of high-throughput experimental approaches and computation methods and at present is limited to a narrow range of materials. The datasets obtained from scattered research studies can make full use of the massive data generated in previous work to solve a comprehensive range of existing scientific issues. With the help of active learning algorithms, data-driven materials design methods can accelerate the progress of development of new materials tremendously.
In the field of Sm-Co-type permanent magnetic materials, the authors have spent over ten years building up a specific database, which contains both experimental and computational data for ~1050 alloys published in the literature over a period of decades. The construction of this database includes three main parts: database models, information management system and database applications. The information management system, known as the “Material Knowledge Information Analysis, Association and Management (MKI-AAM) System”[110], was established to cooperate with the constructed database to improve the efficiency of data collection and the quality of data and also to avoid repetitive data collection. It therefore accelerates the generation of high-quality datasets for machine learning and data-driven materials design.
As shown in Figure 10, the information management system can be divided into three levels: data source, dataset and materials. The one-way arrows indicate that the data sources are research papers, books and scientific reports. The lines without arrows indicate that the two sections are interrelated and the lines with two-way arrows imply that the two sections are not only interrelated, but also are traceable between each other. The data source covers a broad range of literature studies, while the dataset is actually an integrated assemble containing information of the materials in a batch of experiments, including raw materials, preparation technology, test methods, properties, related references and so on. For database applications, in the framework of the information management system, multi-dimensional data retrieval and deep information sharing can be realized. Extract transform load technology[111] was used to obtain datasets for data-driven materials design. In addition, through the development of the application programming interface[112], the information management system can be docked with data analysis software or database exchange platforms of MGE.

Figure 10. Layout of the structure and functions of the “Material Knowledge Information Analysis, Association and Management (MKI-AAM) System” constructed by the authors.
Furthermore, as shown in Figure 11, the MKI-AAM System integrates seven main modules: system settings, document management, dataset management, data statistics, information retrieval, data warehouse and user interface. The core module is the dataset editing, the functions of which include editing and management of the raw materials, preparation process, test methods, material data, related documents, dataset attribute, and so on, as well as the real-time overview of data and warning of repeated data collection.

Figure 11. Example of modules in the construction of the Material Knowledge Information Analysis, Association and Management (MKI-AAM) System.
Figure 12 gives the construction of a MGE-oriented dataset and its inner correlations, generated by the MKI-AAM System using Sm-Co materials as an example. The materials related data are divided into four main parts: process, composition, property and reference. Each part is connected by the “material” label, which records the basic information of the material, so that it can fulfill the requirements of data-driven methods to correlate between materials composition, process, structure and performance. After integrating the relational models of data sources, constituents, phases, structures, preparation processes and properties, the materials data from the data sources can be accurately stored in each relational table of the database, which fully meets the requirements of FAIR (findable, accessible, interoperable, reusable) rules[31,113] for scientific data. Through the above treatment, the data items are closely related and the data system is highly structured, which therefore facilitates the rapid retrieval of different kinds of data and collects them with a standard format.

Figure 12. Construction and inner correlations of a Materials Genome Engineering (MGE)-oriented dataset generated by the Material Knowledge Information Analysis, Association and Management (MKI-AAM) System using Sm-Co materials as an example[110].
The storage status of data in the MGE-oriented database is shown in Figure 13 with a tree diagram using the Sm-Co database as an example. The white boundary is used to separate different datasets and the size of each block represents the quantity of the data in the block. In each dataset, different samples are further separated by the gray boundaries and the data volume of the different properties of each sample is represented by the size of the colored sub-blocks. The visualization of data volume and dataset constituents enables researchers to focus on the key data quickly.

Figure 13. A tree diagram for the data volume and dataset constituents in the Materials Genome Engineering (MGE)-oriented database using the Sm-Co-based systems as an example to show the storage status of data[110].
The data collected in a database inevitably have multi-source heterogeneity. After data preprocessing, a dataset containing sufficient qualified data will be used for data mining and machine learning aimed at materials design. In the constructed MGE-oriented database of Sm-Co type alloys, with the exception of data of materials information, a number of factors that influence the composition, phase constitution, microstructure and properties are also included. Many empirical and semi-empirical formulas used in the modeling studies of materials have been obtained through “trial and error” experimental processes. Although empirical and semi-empirical methods have limitations of lower accuracy and are time consuming, these methods and the resultant criteria provide important references for researchers to set up features of materials for machine learning processes. The concept of “feature engineering”[114] in machine learning has provided materials design with a more comprehensive and accurate approach in massive and multi-dimensional feature space.
Here, machine learning studies on phase stability are demonstrated using the SmCo7-type alloys as an example, which is the most representative group for metastable phase systems in Sm-Co-based permanent magnetic materials. In the modeling process, the correlation between machine learning and materials informatics is analyzed. We first set up five features of the SmCo7-xMx (M is the doping element) alloys for the study of phase stability, i.e., preparation process (Cproc), material form (Cform), grain size (d), doping element type (M) and content (xsup). Thus, the function of SmCo7 phase stability can be described as:
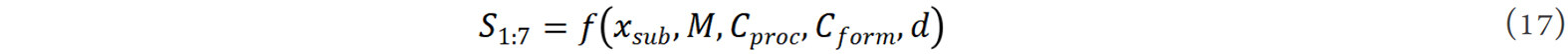
The fundamental features of the elements are then embedded into machine learning models, as listed in Table 1. Some features have a certain linear correlation with each other, so it is necessary to use Pearson correlation coefficient analysis to screen out the representative features without collinearity. Figure 14 shows the data distributions, relationships and Pearson correlation coefficients of the fundamental features, from which some unnecessary features need to be removed.
Fundamental features of elements for machine learning
| Feature name | Symbol | Feature name | Symbol |
|---|---|---|---|
| Atomic number | Z | Electrical conductivity | κ |
| Atomic radius | ra | Heat of fusion | ΔHfus |
| The 1st ionization energy | Ei,1st | Heat of vaporization | ΔHvap |
| Standard atomic weight | Ar | Thermal conductivity | λ |
| Melting point | Tm | Work function | Φ |
| Boiling point | Tb | Electron density | NWS |
| Electronegativity | χ | Atomic volume | Va |

Figure 14. Comprehensive chart of element features: data distributions of each element (histograms in the diagonal), data relationships (lower-left part) and Pearson correlation coefficients (upper-right part).
Finally, seven parameters, including the atomic radius (ra), relative atomic mass (Ar), melting enthalpy (ΔHfus), melting point (Tm), conductivity (κ), first ionization energy (Ei,1st) and electronegativity (χ), are used to represent the characteristics of elements, i.e.,:
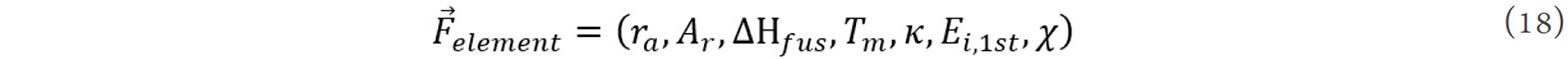
For SmCo7-xMx alloys, the composition feature 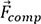 is composed of
is composed of 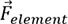 of Sm, Co, doping element M and its content, which can be expressed by:
of Sm, Co, doping element M and its content, which can be expressed by:
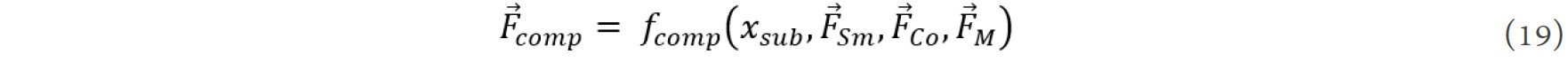
The features for machine learning are constructed by three types of parameters, as given in Table 2.
Classification of features for machine learning
| Formula | Meaning |
|---|---|
xsub·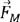 | Product of doping content and element features |
xsub·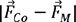 | Absolute difference of element features of M and Co, combined with the doping content |
xsub·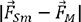 | Absolute difference of element features of M and Sm, combined with the doping content |
The processing of a material (Cproc) and its form (Cproc) are categorical variables and should be converted to numeric (dummy) variables. Thus, the stability of the SmCo7 phase (S1:7) can be expressed as a function of the processing of the material, the form of the material, grain size and compositional feature:
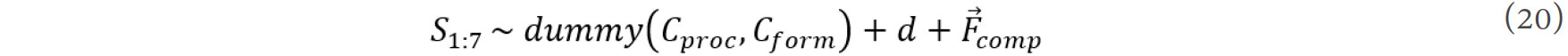
The support vector machine algorithm with a radial basis function as the kernel is selected for machine learning, which has good extrapolation performance. If no element feature is selected as input, the model uses the processing, grain size and material form to evaluate the phase stability and its area under curve (AUC) value is 0.72. If the element feature is selected, the AUC value will be greatly improved, i.e., the accuracy of the prediction is increased. When xsup·Tm and xsup·|χCo-χM| are selected, the AUC value is improved to 0.83 [Figure 15A]. When more features are added, the prediction accuracy cannot be improved obviously anymore.

Figure 15. Machine learning results of phase stability of doped SmCo7 alloys. (A) Average area under curve (AUC) value as a function of the number of composition features, where the highest AUC corresponding to each number of features is marked with red. (B) Data distribution and classification of phase constitutions of SmCo7-xMx alloys in the dataset based on the two selected features of doping elements. (C) Prediction of 1:7 single phase probability with various doping elements for SmCo7-xMx ribbons. (D) Prediction of 1:7 single phase probability with various doping elements for SmCo7-xMx sintered bulks. In (C) and (D), different grain sizes and doping contents are considered for various doping elements.
Figure 15B shows the predicted phase stabilities of the SmCo7-type alloys. Nearly all the 1:7 single-phase alloys are located in the lower left corner in Figure 15B, implying that for the doping elements with a large electronegativity difference or high melting point, to obtain a 1:7 single phase, the suitable range of doping contents will be very narrow. If the elements with a small electronegativity difference and low melting point are selected, there will be a wide range to choose the doping content for stabilizing the 1:7 phase. Based on the machine learning results, the possible elements for doping in Sm-Co magnets are shown in the periodic table [Figure 15C and D]. For each potential doping element, represented by an individual square in the periodic table, the X-axis is the doping content xsup and the Y-axis is the grain size d. The graded color represents the probability of a 1:7 single phase predicted by machine learning. From the comparison between Figure 15C and D, the trend of phase stability for Sm-Co alloys in the form of ribbons is consistent with that of the bulk materials. The difference is that there are more obvious upper and lower limitations of the grain size and doping content in the bulk materials, which may be attributed to the existence of some data from the coarse-grained ingots in the dataset.
As demonstrated above, each individual modeling method has its own features in characterizing the phase stability and transformation criterion. First-principles calculations can predict many intrinsic properties for ideal crystals, regardless of the microstructures and processing of the material. In particular, it can calculate the formation energy and electronic interactions to evaluate the phase stability. However, for phases with complex crystal structures or multicomponent systems with dopants, first-principles calculations are usually difficult to perform with high computational efficiency and reliability. For thermodynamic calculations, the phase with stoichiometry is generally the fundamental component in the model; thus, the phases with varying composition, such as solid solutions and doped matrices, cannot be calculated accurately. Numerical computations based on the cohesive energy are not limited to the type and number of doping elements, but it is difficult to describe the interactions between the atoms of different elements in a compound by the pair potential of independent atoms, especially for multicomponent systems. Data-driven approaches are effective tools to deal with high-dimensional and massive data, but the multi-source heterogeneity of the data and machine learning models with a weak physical background may be significant obstacles for applications. Therefore, we believe that CMS modeling cannot be replaced by data-driven approaches and instead the integration of CMS and materials informatics will strengthen and accelerate progress in the modeling studies of materials science.
Prospects of phase stability studies based on materials informatics
As overviewed in the above sections, modeling studies of phase stability have been developed from traditional computational materials science to materials informatics. With the cooperation of traditional computational methods, database technology and machine learning, significantly accelerated elements screening and materials design are expected to be realized through high-throughput simulations.
In the framework of materials informatics[15], data-driven modeling in materials science is actually to use artificial intelligence to reveal the relationship of composition, structure, processing and properties. The laws behind the data may provide new approaches and perspectives for high-efficiency materials design. Data-driven modeling is a powerful complement and extension of the traditional cognitive paradigm in materials science. To improve the accuracy and efficiency of the modeling, domain knowledge should be introduced into machine learning algorithms. In this respect, the following prospects are proposed.
Database establishment incorporated with data management system
Sufficient materials data represent the basic premise for the implementation of data-driven modeling. The database from which the dataset is extracted should follow the principle of FAIR. With the improvement of data quality, data sharing will become easier and the total social cost will be reduced. At present, the MGE-oriented databases have covered many types of materials, such as superalloys, energy materials, steels, light alloys, composites and rare-earth permanent magnets. It can support the data-driven modeling studies on phase stability in these material systems and promote the high-efficiency development of new materials. With the progress of computational models and methods, numerous computational data can be obtained by high-throughput calculations. However, experimental data are usually scattered, incomplete or not unified in a unit system. In particular, the multi-source heterogeneity of experimental data may cause a serious decline in data quality, which is a large obstacle to data-driven modeling and materials design. Therefore, it is highly desirable to integrate materials science and data processing technologies to acquire knowledge and techniques of data collection, storage, transfer, fusion, and applications. Simultaneously, it is essential to set up standards for data that are suitable for data-driven modeling and materials design. The construction of a data-related information system should be incorporated into the establishment of a database for data-driven applications.
High-throughput calculation models and algorithms
With the improvement of computing capabilities and the functions of integrated computational software, high-throughput calculations will surely be developed with an increasingly high speed. High-throughput calculations are advanced in multi-channel, multi-target and multi-task, and high-concurrency calculations and data management, which are able to generate a large amount of data that will be subsequently used for machine learning. For example, the Materials Project database, which is famous for structural data and the properties of inorganic compounds, has collected data from high-throughput first-principles calculations of ~65,000 materials in the Inorganic Crystal Structure Database. Taking advantages of high-performance computing clusters and developed programs for high-throughput calculations, a variety of new materials have been designed and prepared in experiments[115-117]. However, for some material systems containing complex crystal structures and atomic interactions of multicomponent, even single-task calculation is very time-consuming and high-throughput calculations are challenging to carry out. Therefore, it is highly demanded to develop and optimize models and algorithms for multi-target and multi-task calculations, as well as to combine multi-scale and multi-stage simulation tools, to realize high-efficiency high-throughput calculations.
Interpretable machine-learning-based modeling
Recently, great successes have been achieved in machine learning studies of a large variety of materials[118-120]. However, the lack of interpretability of machine learning algorithms and results limits the applications of machine learning in reality and especially stability-sensitive tasks. Actually, with the development of expression capacity of machine learning algorithms, the models become more complicated and their interpretability may be even weaker. At present, the interpretability of machine learning is still a difficult problem and particularly challenging for researchers in materials science. The models obtained by training based on machine learning algorithms can be considered as a “black box” and are therefore difficult for users to understand the inner working mechanisms.
With the applications of machine learning going deeply into materials design, more attention will be paid to the interpretability of machine learning results. Some valuable attempts have been reported very recently[121,122] concerning the combination of machine learning and first-principles calculations to disclose the effects of alloying elements and propose the strategy of multicomponent materials design. The data-driven modeling on materials phenomena, processing, and relationship of composition, structure and properties are essentially effective accesses to investigate the interpretability of machine learning methods and results. Integration of machine learning with the developed CMS models and material science theories is considered to be the future tendency for developing interpretable machine learning.
Summary
The progress of modeling studies of phase stability in alloys has been reviewed in this article using Sm-Co permanent magnetic materials as examples. The features of various traditional CMS methods for the modeling of phase stability were analyzed and compared, and machine learning studies in the framework of materials informatics were also demonstrated. Both theories and techniques of materials informatics will be developed rapidly by integrating database technologies, high-throughput calculation models and machine learning methods around the core of materials science and engineering. The modeling of phase stability will be significantly advanced by the development of materials informatics, which then accelerates materials design and processing and further progresses the manufacturing of products with high performance.
Declarations
AcknowledgmentsThe authors would like to thank Dr. H. Li (Faculty of Materials and Manufacturing, Beijing University of Technology) for providing CASTEP in partial calculations.
Authors’ contributionsMade substantial contributions to conception and design of this review, writing and editing: Song X
Made substantial contributions to collation of literatures, figures preparation, and writing: Guo K
Performed data analysis, discussion and writing-review: Lu H, Tang F
Performed data acquisition and interpretation: Liu D
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the National Key Program of Research and Development (2018YFB0703902) and the National Natural Science Foundation of China (51631002).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
2. Wu Z, Troparevsky M, Gao Y, Morris J, Stocks G, Bei H. Phase stability, physical properties and strengthening mechanisms of concentrated solid solution alloys. Curr Opin Solid State Mater Sci 2017;21:267-84.
3. Peng HR, Gong MM, Chen YZ, Liu F. Thermal stability of nanocrystalline materials: thermodynamics and kinetics. Int Mater Rev 2016;62:303-33.
6. Seaman L. Development of computational models for microstructural features. AIP Conf Proc 1982;78:118-129.
7. Moriwake H, Kuwabara A, Fisher CA, et al. First-principles calculations of lithium-ion migration at a coherent grain boundary in a cathode material, LiCoO(2). Adv Mater 2013;25:618-22.
8. Derlet P, Dudarev S. Million-atom molecular dynamics simulations of magnetic iron. Prog Mater Sci 2007;52:299-318.
9. Xiao P, Henkelman G. Kinetic monte carlo study of Li intercalation in LiFePO4. ACS Nano 2018;12:844-51.
10. Gránásy L, Tóth GI, Warren JA, et al. Phase-field modeling of crystal nucleation in undercooled liquids - a review. Prog Mater Sci 2019;106:100569.
11. Abe T, Chen Y, Saengdeejimg A, Kobayashi Y. Computational phase diagrams for the Nd-based magnets based on the combined ab initio/CALPHAD approach. Scripta Materialia 2018;154:305-10.
13. Wang J, Yang T, Zorn JA, et al. Strain anisotropy and magnetic domain structures in multiferroic heterostructures: High-throughput finite-element and phase-field studies. Acta Materialia 2019;176:73-83.
14. Wang P, Sun S, Zhang Q, Zhang TY. A brief introduction of mechanoinformations. Chin J Nat 2018;40:313-22. (in Chinese)
16. Xiong J, Shi S, Zhang T. A machine-learning approach to predicting and understanding the properties of amorphous metallic alloys. Materials & Design 2020;187:108378.
17. Xiong J, Shi S, Zhang T. Machine learning prediction of glass-forming ability in bulk metallic glasses. Comp Mater Sci 2021;192:110362.
18. Xiong J, Zhang T, Shi S. Machine learning prediction of elastic properties and glass-forming ability of bulk metallic glasses. MRS Communications 2019;9:576-85.
19. Wei Q, Xiong J, Sun S, Zhang T. Multi-objective machine learning of four mechanical properties of steels. Sci Sin-Tech 2021;51:722-36.
20. Zhang T, He Y, Wang J, Sun S. Machine learning prediction of the hardness of tool and mold steels. Sci Sin-Tech 2019;49:1148-58.
21. Wang WY, Zhang Y, Liaw PK. Editorial: data-driven integrated computational materials engineering for high-entropy materials. Front Mater 2021;8:664829.
22. Zou C, Li J, Wang WY, et al. Integrating data mining and machine learning to discover high-strength ductile titanium alloys. Acta Materialia 2021;202:211-21.
23. Li R, Xie L, Wang WY, Liaw PK, Zhang Y. High-throughput calculations for high-entropy alloys: a brief review. Front Mater 2020;7:290.
24. Yi Wang W, Li J, Liu W, Liu Z. Integrated computational materials engineering for advanced materials: a brief review. Comp Mater Sci 2019;158:42-8.
25. Wang H, Xiang X, Zhang L. On the data-driven materials innovation infrastructure. Engineering 2020;6:609-11.
26. de Pablo JJ, Jackson NE, Webb MA, et al. New frontiers for the materials genome initiative. npj Comput Mater 2019;5.
27. de Pablo JJ, Jones B, Kovacs CL, Ozolins V, Ramirez AP. The materials genome Initiative, the interplay of experiment, theory and computation. Curr Opin Solid State Mater Sci 2014;18:99-117.
28. Himanen L, Geurts A, Foster AS, Rinke P. Data-driven materials science: status, challenges, and perspectives. Adv Sci 2019;6:1900808.
29. Wang H, Xiang Y, Xiang XD, Chen LQ. Materials genome enables research and development revolution. Sci Tech Rev 2015;33:13-9. (in Chinese)
30. Shi SQ, Xu JW, Cui YH, et al. Multiscale materials computational methods. Sci Tech Rev 2015;33:20-30. (in Chinese)
31. Wang H, Xiang XD, Zhang LT. Data + AI: the core of materials genomic engineering. Sci Tech Rev 2018;36:15-21. (in Chinese)
32. Su YJ, Fu HD, Bai Y, Jiang X, Xie JX. Progress in materials genome engineering in China. Acta Metall Sin 2020;56:1313-23. (in Chinese)
33. Wang WY, Li P, Lin D, et al. DID code: a bridge connecting the materials genome engineering database with inheritable integrated intelligent manufacturing. Engineering 2020;6:612-20.
34. Wang WY, Tang B, Lin D, et al. A brief review of data-driven ICME for intelligently discovering advanced structural metal materials: insight into atomic and electronic building blocks. J Mater Res 2020;35:872-89.
35. Chen C, Zuo Y, Ye W, Li X, Deng Z, Ong SP. A critical review of machine learning of energy materials. Adv Energy Mater 2020;10:1903242.
36. Yang F, Li Z, Wang Q, et al. Cluster-formula-embedded machine learning for design of multicomponent β-Ti alloys with low Young’s modulus. npj Comput Mater 2020;6:101.
37. Möller JJ, Körner W, Krugel G, Urban DF, Elsässer C. Compositional optimization of hard-magnetic phases with machine-learning models. Acta Materialia 2018;153:53-61.
38. Jinnouchi R, Asahi R. predicting catalytic activity of nanoparticles by a DFT-aided machine-learning algorithm. J Phys Chem Lett 2017;8:4279-83.
39. Ling J, Chen W, Sheng Y, Li W, Zhang L, Du Y. A MGI-oriented investigation of the Young’s modulus and its application to the development of a novel Ti-Nb-Zr-Cr bio-alloy. Mater Sci Eng C Mater Biol Appl 2020;106:110265.
40. Liu Y, Zhao T, Ju W, Shi S. Materials discovery and design using machine learning. Journal of Materiomics 2017;3:159-77.
42. Liu S, Su Y, Yin H, et al. An infrastructure with user-centered presentation data model for integrated management of materials data and services. npj Comput Mater 2021;7:88.
43. Yılmaz B, Yıldırım R. Critical review of machine learning applications in perovskite solar research. Nano Energy 2021;80:105546.
44. Ludwig A. Discovery of new materials using combinatorial synthesis and high-throughput characterization of thin-film materials libraries combined with computational methods. npj Comput Mater 2019;5.
45. Gutfleisch O, Willard MA, Brück E, Chen CH, Sankar SG, Liu JP. Magnetic materials and devices for the 21st century: stronger, lighter, and more energy efficient. Adv Mater 2011;23:821-42.
46. Song X, Zhang Z, Lu N, et al. Crystal structures and magnetic performance of nanocrystalline Sm-Co compounds. Front Mater Sci 2012;6:207-15.
47. Zhang Z, Song X, Qiao Y, et al. A nanocrystalline Sm-Co compound for high-temperature permanent magnets. Nanoscale 2013;5:2279-84.
48. Xu W, Song X, Lu N, Huang C. Thermodynamic and experimental study on phase stability in nanocrystalline alloys. Acta Materialia 2010;58:396-407.
49. Zhang Z, Song X, Xu W. Phase evolution and its effects on the magnetic performance of nanocrystalline SmCo7 alloy. Acta Materialia 2011;59:1808-17.
50. Luo J, Liang JK, Rao GH. Formation, crystal structure and magnetic properties of rare-earth transition-metal intermetallic compounds (I). J Chin Rare Earth Soc 2012;30:385-402. (in Chinese)
51. Luo J, Liang JK, Rao FH. Formation, crystal structure and magnetic properties of rare-earth transition-metal intermetallic compounds (II). J Chin Rare Earth Soc 2012;30:513-24. (in Chinese)
52. Shen B, Yu C, Baker AA, et al. Chemical synthesis of magnetically hard and strong rare earth metal based nanomagnets. Angew Chem Int Ed Engl 2019;58:602-6.
53. Li X, Lou L, Song W, et al. Novel bimorphological anisotropic bulk nanocomposite materials with high energy products. Adv Mater 2017;29:1606430.
55. Mccallum R, Lewis L, Skomski R, Kramer M, Anderson I. Practical aspects of modern and future permanent magnets. Annu Rev Mater Res 2014;44:451-77.
57. Liu XM, Liu GQ, Li DP, Wang HB, Song XY. Preparation and properties of polycrystalline and nanocrystalline Sm3Co alloys. Acta Phys Sin 2014;63:098102. (in Chinese)
58. Li D, Song X, Zhang Z, Lu N, Qiao Y, Liu X. Preparation and properties of single-phase Sm5Co2 nanocrystalline alloy. Acta Metallurgica Sinica 2012;48:1248.
59. Song X, Lu N, Seyring M, et al. Abnormal crystal structure stability of nanocrystalline Sm2Co17 permanent magnet. Appl Phys Lett 2009;94:023102.
60. Lu N, Song X, Zhang J. Crystal structure and magnetic properties of ultrafine nanocrystalline SmCo3 compound. Nanotechnology 2010;21:115708.
61. Zhang Z, Song X, Xu W, Seyring M, Rettenmayr M. Crystal structure and magnetic performance of single-phase nanocrystalline SmCo7 alloy. Scripta Materialia 2010;62:594-7.
62. Zhang Z, Song X, Xu W, Li D, Liu X. Crystal structure and magnetic performance of nanocrystalline SmCo9.8 alloy. J Appl Phys 2011;110:124318.
63. Yue M, Zhang X, Liu JP. Fabrication of bulk nanostructured permanent magnets with high energy density: challenges and approaches. Nanoscale 2017;9:3674-97.
64. Xu WW, Song XY, Zhang ZX. multiphase equilibrium, phase stability and phase transformation in nanocrystalline alloy systems. NANO 2012;07:1250012.
65. Mchale JM. Surface energies and thermodynamic phase stability in nanocrystalline aluminas. Science 1997;277:788-91.
66. Ikeda Y, Grabowski B, Körmann F. Ab initio phase stabilities and mechanical properties of multicomponent alloys: a comprehensive review for high entropy alloys and compositionally complex alloys. Materials Characterization 2019;147:464-511.
67. Xu WW, Song XY, Li ED, Wei J, Li LM. Phase configuration and stability in the nanocrystalline Sm-Co system. Acta Phys Sin 2009;58:3280-6. (in Chinese)
68. Zhang H, Banfield JF. Thermodynamic analysis of phase stability of nanocrystalline titania. J Mater Chem 1998;8:2073-6.
69. Song X, Zhang J, Li L, Yang K, Liu G. Correlation of thermodynamics and grain growth kinetics in nanocrystalline metals. Acta Materialia 2006;54:5541-50.
70. Guinea F, Rose JH, Smith JR, Ferrante J. Scaling relations in the equation of state, thermal expansion, and melting of metals. Appl Phys Lett 1984;44:53-5.
71. Xu W, Song X, Zhang Z. Thermodynamic study on metastable phase: from polycrystalline to nanocrystalline system. Appl Phys Lett 2010;97:181911.
72. Xu WW, Song XY, Li ED, Wei J, Zhang JX. Thermodynamic study on phase stability in nanocrystalline Sm-Co alloy system. J Appl Phys 2009;105:104310.
73. Xu W, Song X, Zhang Z, Liang H. Experimental and modeling studies on phase stability of nanocrystalline magnetic Sm2Co7. Materials Science and Engineering: B 2013;178:971-6.
74. Xu W, Song X, Lu N, Seyring M, Rettenmayr M. Nanoscale thermodynamic study on phase transformation in the nanocrystalline Sm2Co17 alloy. Nanoscale 2009;1:238-44.
75. Meng Q, Zhou N, Rong Y, Chen S, Hsu T, Zuyao X. Size effect on the Fe nanocrystalline phase transformation. Acta Materialia 2002;50:4563-70.
76. Hu B, Zhang HQ, Zhang J, Yang MJ, Du Y, Zhao DD. Progress in interfacial thermodynamics and grain boundary complexion diagram. Acta Metall Sinica 2021;57:1199-214.
77. Song X, Zhang J, Yue M, et al. Technique for preparing ultrafine nanocrystalline bulk material of pure rare-earth metals. Adv Mater 2006;18:1210-5.
78. Fecht HJ. Intrinsic instability and entropy stabilization of grain boundaries. Phys Rev Lett 1990;65:610-3.
79. Wagner M. Structure and thermodynamic properties of nanocrystalline metals. Phys Rev B Condens Matter 1992;45:635-9.
80. Yang CC, Li S. Size-dependent phase stability of silver nanocrystals. J Phys Chem C 2008;112:16400-4.
82. Guo K, Lu H, Mao F, et al. How non-ferromagnetic Mn enhances the magnetization of SmCo7 based alloys. Nanoscale 2020;12:5567-77.
83. Yao Z, Jiang C. Structure and magnetic properties of SmCoxTi0.4-1:7 ribbons. J Magn Magn Mater 2008;320:1073-7.
84. Zhou J, Skomski R, Chen C, Hadjipanayis GC, Sellmyer DJ. Sm-Co-Cu-Ti high-temperature permanent magnets. Appl Phys Lett 2000;77:1514-6.
85. Luo J, Liang JK, Guo YQ, et al. Crystal structure and magnetic properties of SmCo5.85Si0.90 compound. Appl Phys Lett 2004;84:3094-6.
86. Guo Y, Li W, Feng W, et al. Structural stability and magnetic properties of SmCo7-xGax. Appl Phys Lett 2005;86:192513.
87. Luo J, Liang J, Guo Y, et al. Effects of the doping element on crystal structure and magnetic properties of Sm(Co,M)7 compounds (M=Si, Cu, Ti, Zr, and Hf). Intermetallics 2005;13:710-6.
88. Al-omari I, Zhou J, Sellmyer D. Magnetic and structural properties of SmCo6.75-xFexZr0.25 compounds. J Alloys Compd 2000;298:295-8.
89. Huang MQ, Wallace WE, McHenry M, Chen Q, Ma BM. Structure and magnetic properties of SmCo7-xZrx alloys (x = 0-0.8). J Appl Phys 1998;83:6718-20.
90. Zhou J, Al-omari IA, Liu JP, Sellmyer DJ. Structure and magnetic properties of SmCo7-xTix with TbCu7-type structure. J Appl Phys 2000;87:5299-301.
91. Luo J, Liang JK, Guo YQ, et al. Crystal structure and magnetic properties of SmCo7-xHfx compounds. Appl Phys Lett 2004;85:5299-301.
92. Guo Y, Feng W, Li W, Luo J, Liang J. Magnetism and phase stability of R(Co,M)7 pseudobinary intermetallics with TbCu7-type structure. J Appl Phys 2007;101:023919.
93. Li Y, Shen J, Chen Y. Atomistic simulation for disordered TbCu7-type compounds SmCo7 and Sm(Co,T)7 (T=Ti, Ga, Si, Cu, Hf, Zr). Solid State Sciences 2010;12:33-8.
94. Sobolev A, Golovnia O, Popov A. Embedded atom potential for Sm-Co compounds obtained by force-matching. J Magn Magn Mater 2019;490:165468.
95. Song XY, Xu WW, Zhang ZX. Nanoscale stabilization of metastable phase: thermodynamic model and experimental studies. Acta Phys Sin 2012;61:200510. (in Chinese)
96. Seyring M, Song X, Zhang Z, Rettenmayr M. Concurrent ordering and phase transformation in SmCo7 nanograins. Nanoscale 2015;7:12126-32.
97. Hua G, Song X, Tang F, et al. Modeling and experimental studies of Hf-doped nanocrystalline SmCo7 alloys. Cryst Eng Comm 2016;18:8080-8.
98. Mao F, Lu H, Liu D, Guo K, Tang F, Song X. Structural stability and magnetic properties of SmCo5 compounds doped with transition metal elements. J Alloys Compd 2019;810:151888.
99. Hua G, Song X, Liu D, Wang D, Wang H, Liu X. Effects of Hf on phase structure and magnetic performance of nanocrystalline SmCo7-type alloy. J Mater Sci 2016;51:3390-7.
100. Das B, Choudhary R, Skomski R, et al. Anisotropy and orbital moment in Sm-Co permanent magnets. Phys Rev B 2019;100:024419.
101. Söderlind P, Landa A, Locht ILM, et al. Prediction of the new efficient permanent magnet SmCoNiFe3. Phys Rev B 2017;96:100404.
102. Gavrikov IS, Karpenkov DY, Zheleznyi MV, Kamynin AV, Khotulev ES, Bazlov AI. Effect of Ni doping on stabilization of Sm(Co1-xFex)5 compound: thermodynamic calculation and experiment. J Phys Condens Matter 2020;32:425803.
103. Chen Nx, Ren Gb. Carlsson-Gelatt-Ehrenreich technique and the Möbius inversion theorem. Phys Rev B Condens Matter 1992;45:8177-80.
104. Chen Nx. Modified Möbius inverse formula and its applications in physics. Phys Rev Lett 1990;64:1193-5.
105. Shen J, Qian P, Chen N. Atomistic investigation on site preference and lattice vibrations of Sm(Co,M)12 (M = Cr, Ti, V, Nb, Fe). J Alloys Compd 2005;388:208-14.
106. Shen J, Qian P, Chen N. Atomistic simulation on phase stability and site preference of R2 (Co, Mn)17 (R = Nd, Sm, Gd). Modelling Simul Mater Sci Eng 2005;13:239-47.
107. Wang AY, Murdock RJ, Kauwe SK, et al. Machine learning for materials scientists: an introductory guide toward best practices. Chem Mater 2020;32:4954-65.
108. Ong SP. Accelerating materials science with high-throughput computations and machine learning. Comp Mater Sci 2019;161:143-50.
109. Liu D, Guo K, Tang F, et al. Selecting doping elements by data mining for advanced magnets. Chem Mater 2019;31:10117-25.
110. Liu X, Song X, Guo K, Liu D, Mao F. Development of database and information management system for data-driven materials design. Sci Sin-Tech 2020;50:786-800.
111. Vassiliadis P. A survey of extract-transform-load technology. Int J Data Warehous Min 2009;5:1-27.
112. Ong SP, Cholia S, Jain A, et al. The materials application programming interface (API): a simple, flexible and efficient API for materials data based on REpresentational State Transfer (REST) principles. Comp Mater Sci 2015;97:209-15.
113. Wilkinson MD, Dumontier M, Aalbersberg IJ, et al. The FAIR guiding principles for scientific data management and stewardship. Sci Data 2016;3:160018.
114. Dai D, Xu T, Wei X, et al. Using machine learning and feature engineering to characterize limited material datasets of high-entropy alloys. Comp Mater Sci 2020;175:109618.
115. Jain A, Ong SP, Hautier G, et al. Commentary: The Materials Project: a Materials genome approach to accelerating materials innovation. APL Materials 2013;1:011002.
116. Kim E, Huang K, Saunders A, Mccallum A, Ceder G, Olivetti E. Materials synthesis insights from scientific literature via text extraction and machine learning. Chem Mater 2017;29:9436-44.
117. Wu Y, Lazic P, Hautier G, Persson K, Ceder G. First principles high throughput screening of oxynitrides for water-splitting photocatalysts. Energy Environ Sci 2013;6:157-68.
118. Schmidt J, Shi J, Borlido P, Chen L, Botti S, Marques MAL. Predicting the thermodynamic stability of solids combining density functional theory and machine learning. Chem Mater 2017;29:5090-103.
119. Tao Q, Xu P, Li M, Lu W. Machine learning for perovskite materials design and discovery. npj Comput Mater 2021;7:1-18.
120. Durodola J. Machine learning for design, phase transformation and mechanical properties of alloys. Prog Mater Sci 2021; doi: 10.1016/j.pmatsci.2021.100797.
121. Lu Z, Chen X, Liu X, et al. Interpretable machine-learning strategy for soft-magnetic property and thermal stability in Fe-based metallic glasses. npj Comput Mater 2020;6:1-9.
Cite This Article
How to Cite
Song, X.; Guo K.; Lu H.; Liu D.; Tang F. Integrating computational materials science and materials informatics for the modeling of phase stability. J. Mater. Inf. 2021, 1, 7. http://dx.doi.org/10.20517/jmi.2021.06
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.