


Role of plasminogen activator inhibitor-1 (PAI-1) in age-related cardiovascular pathophysiology
 0
0 
Abstract
Cardiovascular aging underpins the development of age-related diseases, including heart failure and vascular dysfunction, and is driven by molecular and cellular mechanisms described in the hallmarks of aging. Plasminogen activator inhibitor-1 (PAI-1), a key regulator of fibrinolysis, also mediates processes like vascular stiffness, cellular senescence, and immune evasion. This review highlights PAI-1’s role in cardiovascular aging with a special emphasis on senescence, a key hallmark of aging. It further explores PAI-1’s therapeutic potential, with a focus on its contribution to ECM remodeling, senescence signaling, and immune checkpoint regulation. Targeting PAI-1 could provide a promising strategy to mitigate age-related cardiovascular disease.

Keywords
INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of death worldwide and is closely linked to aging[1]. As the global population over 60 is expected to double from 1 billion in 2020 to 2.1 billion by 2050[2], addressing age-related cardiovascular conditions becomes increasingly urgent. The geroscience hypothesis posits that aging drives not only CVD but also other chronic diseases, suggesting that interventions targeting aging processes could mitigate these conditions[3]. Among these processes, the hallmarks of aging - such as genomic instability, cellular senescence, and disrupted proteostasis - provide a framework to dissect molecular and cellular changes that impair cardiovascular resilience, manifesting as hypertension, vascular stiffness, and heart failure[4,5].
Plasminogen activator inhibitor-1 (PAI-1), a serine protease inhibitor, is closely linked to the hallmarks of aging and aging-related CVD[6]. PAI-1 was originally identified as the principal inhibitor of tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA), and PAI-1 thereby regulates fibrinolysis and extracellular matrix (ECM) remodeling[7]. Subsequent studies revealed additional t-PA targets that are impacted by PAI-1 inhibition and contribute to cardiovascular aging, such as fibroblast growth factor 23 (FGF23)[8], brain-derived neurotrophic factor (BDNF)[9,10], insulin-like growth factor binding protein 3 (IGFBP3)[11], and matrix metalloproteinase-9 (MMP-9)[12]. PAI-1 is primarily secreted by endothelial cells, hepatocytes, and adipocytes, and it is cleared by the liver with a half-life of approximately
VASCULAR STIFFNESS
Vascular stiffness overview
Vascular stiffness accelerates the development of CVD, especially in aging populations[20]. As arteries stiffen due to reduced elastin, increased collagen deposition, and calcification, they lose elasticity and become less responsive to changes in blood flow[21]. This pathophysiology leads to increased systolic blood pressure and pulse pressure, which raise left ventricular pressure; increased left ventricular pressure causes left ventricular hypertrophy that can progress to heart failure. Stiff arteries also fail to cushion the pulsatile flow of blood, damaging the microvasculature in organs like the brain, kidneys, and heart. Microvascular damage contributes to the development of hypertension and accelerates atherosclerosis, which together increase the risk of heart attacks and strokes[22]. At the physiological level, pulse wave velocity (PWV) is a key measure of arterial stiffness. PWV assesses the pulse wave transit time across a fixed distance, typically between the carotid and femoral arteries; the higher the velocity, the stiffer the vessel. PWV increases with age, particularly in the distal aorta[23]. Similarly, there is an associated increase in the diameter of the aorta with advancing age[24].
At the molecular level, several pathways contribute to vascular stiffening. ECM composition changes with age, specifically with increased collagen deposition and elastin fiber degradation[25]. Furthermore, increased ECM stiffness due to increased collagen and decreased elastin was shown to promote transdifferentiation of fibroblasts to myofibroblasts[26,27]. Activated myofibroblasts secrete additional ECM components and exacerbate the stiffness of the vasculature[28]. Endothelial cells also modulate vascular tone and vasodilation through the release of nitric oxide (NO). Endothelial NO synthase (eNOS) produces NO, which diffuses to neighboring vascular smooth muscle cells (VSMCs). In VSMCs, NO activates soluble guanylate cyclase, increasing cyclic guanosine monophosphate (cGMP) levels, which in turn activate protein kinase G (PKG). PKG inhibits voltage-gated calcium channels, reducing calcium influx and leading to vasodilation[29]. NO production was also shown to negatively regulate VSMC collagen production[30].
PAI-1 contribution to vascular stiffness
PAI-1 is a serine protease inhibitor (SERPIN), encoded by the SERPINE1 gene, that was originally shown to reduce fibrinolysis through its inhibition of t-PA and u-PA[7]. Later work revealed that PAI-1 inhibition of t-PA and u-PA could contribute to ECM protein accumulation and increased vascular stiffness[31]. The uPA/tPA/plasmin/MMP proteolytic system regulates ECM turnover and homeostasis[32]. Since PAI-1 acts as an upstream inhibitor of uPA and tPA, it thus influences the tPA and uPA targets of plasmin and MMP activities, which degrade ECM components[33,34]. As a result, elevated PAI-1 levels are implicated in tissue fibrosis, driven by reduced ECM degradation[35] and altered cellular migration[36,37] [Figure 1A].
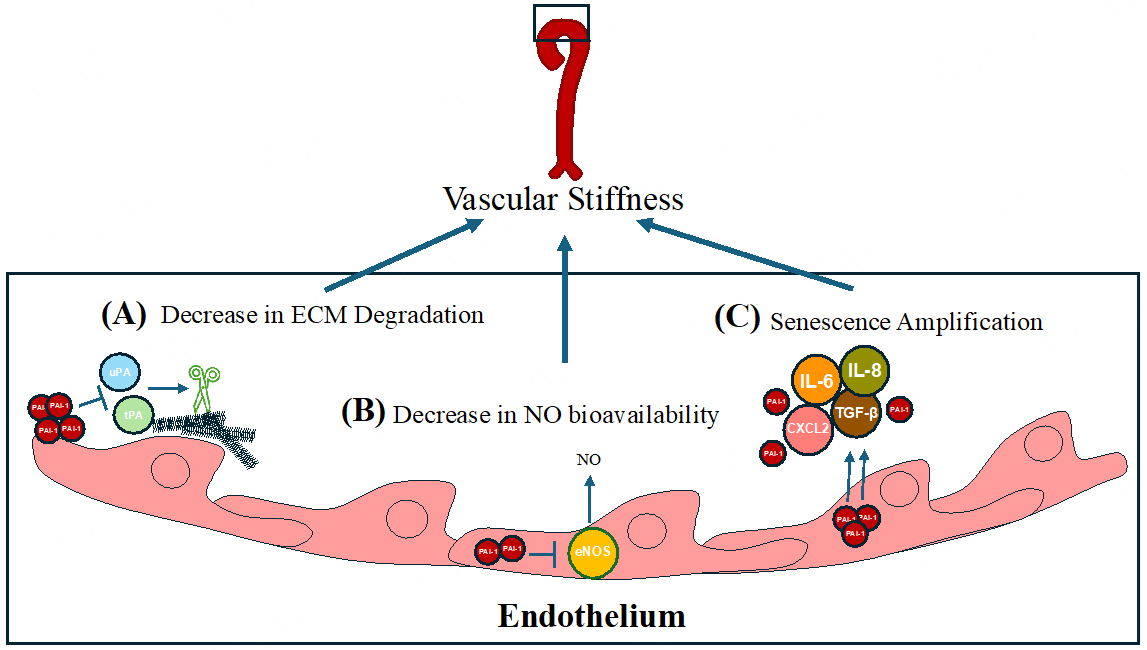
Figure 1. The role of PAI-1 in vascular stiffness and senescence amplification. (A) PAI-1 inhibits degradation of the ECM via inhibiting the tPA/uPA-mediated cleavage of metalloproteinases. (B) PAI-1 inhibits eNOS, reducing NO bioavailability and promoting vascular stiffness in the endothelium. (C) PAI-1 amplifies senescence by increasing SASP factors (IL-6, IL-8, TGF-β, CXCL2), which exacerbate vascular dysfunction and aging.
Subsequent studies identified novel molecular mechanisms by which PAI-1 might contribute to stiffness independently of its protease inhibitor activity. For example, L-NAME inhibits eNOS and increases blood pressure due to reduced NO availability; inhibition of PAI-1 was shown to blunt L-NAME-induced increase in blood pressure in mice[38]. Garcia et al. demonstrated that PAI-1 directly binds to eNOS, inhibits its activity in cultured endothelial cells, and that this interaction is ablated by using SERPINE1 siRNA[39]; in vivo studies will determine whether this mechanism operates in the vasculature. Additionally, eNOS inhibition by PAI-1 might increase intracellular reactive oxygen species (ROS), potentially leading to cellular senescence (see below). This potential pathway is summarized in Figure 1B. Finally, recent atomic force microscopy studies in cultured VSMCs revealed that PAI-1 contributed to stiffness through increased F-actin polymerization; pharmacological PAI-1 inhibition or Serpine1 siRNA knockdown reduced intrinsic cellular stiffness, suggesting a mechanism independent of collagen and elastin content[40]. These studies suggest a model where increased PAI-1 decreases NO bioavailability and increases VSMC stiffness via F-actin polymerization.
SENESCENCE
Senescence overview
Building on PAI-1’s role in vascular stiffness, its influence extends to cellular senescence, a hallmark of aging that further impairs vascular function and accelerates cardiovascular pathology. Specifically, senescent endothelial cells exhibit lower eNOS expression, linking PAI-1, NO bioavailability, and vascular aging[41]. Cellular senescence occurs in response to stressors like DNA damage, oxidative stress, and telomere shortening - all of which increase with age[42]. In the cardiovascular system, endothelial cells experience constant mechanical stress from blood flow, leading to high turnover, and a consequent telomeric shortening, which increases susceptibility to senescence[43]. The endothelium is vital for maintaining appropriate vascular tone, and the accumulation of senescent cells impairs vascular function[44]. For example, senescent cell accumulation in the aorta has been associated with pathologies such as aortic aneurysm[45]. At the molecular level, senescent cells upregulate the p53-p21 and/or RB-p16 pathways, blocking the cell cycle[46]. Although they do not proliferate, senescent cells remain metabolically active and secrete senescence-associated secretory phenotype (SASP) factors; besides PAI-1 itself, the SASP includes pro-inflammatory cytokines, growth factors, and proteases that induce senescence in neighboring cells. Senescent cells negatively affect the vasculature by impairing the regenerative capacity of endothelial and smooth muscle cells, disrupting tissue repair, and leading to ECM protein accumulation, all of which promote vascular stiffening and dysfunction[47].
Current approaches to reducing senescent cell burden involve the induction of apoptosis of senescent cells (senolytic) or neutralizing SASP factors (senomorphic). For instance, targeting p16-positive cells for apoptosis extended median lifespan in mice by up to 25%[48-50]. Although these studies show promising results, the initial studies use genetically engineered mice with inducible caspase activation under the p16 promoter, posing challenges for direct clinical translation. The combination of dasatinib and quercetin
PAI-1 contribution to senescence
Multiple studies have demonstrated that PAI-1 directly contributes to senescence. Excess PAI-1 is associated with an increase in SASP factors such as IL-6, IL-1β, IL-8, CXCL2, and TGFβ[54]. Mechanistically, early reports showed that PAI-1 overexpression induced senescence in p53-deficient fibroblasts, indicating PAI-1 signals independently of p53[55]. Our group and others have previously reviewed PAI-1's role in marking and inducing senescence[54,56-58]. Briefly, PAI-1 is synthesized and secreted by senescent cells, contributing directly to the development of cellular senescence downstream of p53 and upstream of insulin-like growth factor binding protein-3 (IGFBP-3). IGFBP-3 levels increase in senescent cells and directly stimulate senescence. t-PA mediates the proteolytic inactivation of IGFBP-3, while PAI-1 prevents this process, thereby promoting senescence.
Genetic PAI-1 deficiency and pharmacological PAI-1 inhibition reduce senescent cell markers in vitro and in vivo. In mice exposed to L-NAME, treatment with PAI-1 inhibitor TM5441 prevented increased senescence markers like p16 expression and telomere shortening in the aorta of L-NAME-treated mice[38]. In addition, PAI-1 inhibition in endothelial cells protected against homocysteine-induced senescence, preventing increased SA-β-Gal while enhancing NRF2 levels[59]. Similar protective effects against senescence were observed in murine cardiomyocytes exposed to doxorubicin[60]. Interestingly, treatment with
Other than contributing to the aging phenotype, senescence plays a crucial role in tumor-protection mechanisms; hence, a concern with senolytic and senomorphic therapies is the uncertainty of long-term use[46]. Possible mechanisms underlying the alterations in senescent cells along the vasculature are summarized in Figure 1C. The involvement of PAI-1 in cancer leads to a broader perspective on how PAI-1 traverses the complex landscape of aging, intertwining with processes such as senescence, immune modulation, and cancer progression.
EVADING IMMUNE SURVEILLANCE
Beyond its direct effects on stiffness and senescence, PAI-1 also modulates immune responses, linking its overexpression to immune evasion mechanisms that exacerbate vascular aging. Released senescence-associated secretory phenotype (SASP) factors, including pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α, recruit inflammatory cells to the vasculature, contributing to a low-grade inflammation termed “inflammaging”[61]. This chronic inflammation feeds into a cycle of cellular senescence, endothelial dysfunction, and arterial stiffening, accelerating pathologies like atherosclerosis that are central to cardiovascular aging[44].
Recent studies have identified a connection between the immune checkpoint inhibitor Programmed Death Ligand-1 (PD-L1) and cellular senescence, with implications extending to both cardiovascular and cancer contexts. PD-L1, traditionally known for enabling cancer cells to evade immunosurveillance by binding to Programmed Cell Death Protein-1 (PD-1) on T cells and inducing T cell exhaustion[62], is also upregulated in senescent fibroblasts following exposure to SASP factors via the JAK1/STAT3 pathway[63]. This upregulation allows senescent cells to evade clearance by CD8+ T cells, a mechanism paralleling immune evasion in tumors. Blocking the PD-1/PD-L1 interaction has been shown to reduce senescence markers
PAI-1 appears to bridge these processes by upregulating PD-L1 expression in both tumor and senescent cells, mediated through the PAI-1/uPAR/LRP-1 complex and JAK1/STAT3 activation[69]. This upregulation may enable senescent cells in the vasculature to escape immune surveillance, contributing to the accumulation of dysfunctional cells that drive CV aging. Similarly, in cancer, PAI-1’s role in promoting metastasis - such as increasing ECM stiffness via integrinβ1-induced collagen I branching in endometrial tumor models[70,71] - mirrors its effects on vascular stiffening in cardiovascular disease. The release of PAI-1 into the bloodstream with aging may thus create a pro-tumorigenic microenvironment while simultaneously exacerbating vascular inflammation and stiffness, linking cancer progression to cardiovascular pathology[72-74].
The overlap between cancer and senescence is further evident in their shared secretory phenotypes, which release factors promoting inflammation and tissue remodeling[73]. While senescence is generally tumor-suppressive by halting proliferation, the accumulation of senescent cells in aged cardiovascular tissues can foster a pro-tumorigenic environment through SASP factors like PAI-1[75]. This duality suggests that age-related increases in circulating PAI-1 could amplify both cardiovascular dysfunction (atherosclerosis, hypertension) and cancer risk (metastasis), particularly in the context of vascular inflammation and ECM remodeling. For instance, PAI-1’s repression of adhesion genes via LRP-1 internalization may contribute to both senescence-associated tissue remodeling and cancer cell detachment during metastasis[72], a process that could parallel vascular matrix changes in cardiovascular aging. Future research should elucidate the molecular mechanisms determining PAI-1’s context-specific effects, as well as explore whether PAI-1 inhibition could simultaneously mitigate vascular senescence and tumor progression. This dual-targeting potential could offer a novel therapeutic avenue for addressing the multisystem decline associated with aging, with particular relevance to cardiovascular disease prevention and management.
PLEIOTROPIC ACTIONS OF PAI-1 AND OUTLOOK
While PAI-1 drives cardiovascular aging through stiffness, senescence, and immune modulation, its pleiotropic effects span additional physiological systems, broadening its therapeutic relevance.
Metabolism and dyslipidemia
With increasing age, the risk and impact of dyslipidemia on cardiovascular health increase significantly. Aging is associated with changes in lipid metabolism, such as higher LDL cholesterol and triglycerides, and a decline in HDL cholesterol levels[29]. These lipid imbalances, combined with age-related changes in blood vessels - including reduced elasticity and increased arterial stiffness - create a "perfect storm" for atherosclerosis.
Notably, PAI-1's role extends into the interplay between dyslipidemia and aging, exacerbating the risk of CVD[76]. Hyperinsulinemia, a hallmark of insulin resistance in aging and metabolic syndrome, is associated with amplified PAI-1 levels. Nordt et al. demonstrated that proinsulin and insulin induce PAI-1 expression in rabbits[77]. It has recently been reported that t-PA binds to microsomal triglyceride transfer protein (MTP) to prevent apolipoprotein B (ApoB) lipidation in hepatocytes[78]. Increased PAI-1 sequesters t-PA from MTP, promoting ApoB lipidation and very-low-density lipoprotein (VLDL) assembly[78]. This insulin-mediated PAI-1 activation exacerbates dyslipidemia by promoting VLDL assembly and impairs fibrinolysis, creating a feedback loop that accelerates atherosclerosis and cardiovascular risk. Additionally, treatment of mice with the PAI-1 inhibitor TM5614 was shown to reduce proprotein convertase subtilisin/kexin type 9 (PCSK9) expression while increasing fibroblast growth factor 21 (FGF21)[79,80]. High PCSK9 levels reduce LDL receptor availability, increasing blood cholesterol; therefore, PAI-1 inhibition might decrease circulating cholesterol by lowering PCSK9 levels. These mechanisms altering metabolism are summarized in Figure 2.

Figure 2. PAI-1’s role in dyslipidemia and metabolic dysfunction. Hyperinsulinemia elevates PAI-1 levels, which inhibit t-PA interaction with MTP, promoting VLDL assembly in hepatocytes. PAI-1 also upregulates PCSK9 and FGF21, exacerbating dyslipidemia and cardiovascular risk.
Currently, caloric restriction is the “gold standard” in preventing metabolic dysfunction[81] and circulating PAI-1 levels are reduced following caloric restriction[82]. Specifically, during caloric restriction, liver-derived PAI-1 decreases[83]. This reduction in PAI-1 was associated with increased plasmin activity on muscle satellite stem cells and their expansion after injury[83]. Muscle atrophy during aging is linked to metabolic disorders due to impaired insulin sensitivity, reduced mitochondrial function, and chronic low-grade inflammation, which collectively disrupt protein synthesis and muscle regeneration[84]. These metabolic imbalances, exacerbated by elevated PAI-1 levels, suggest a mechanism by which PAI-1 accelerates the loss of muscle mass and strength, contributing to frailty and increased risk of metabolic diseases such as type 2 diabetes. Altogether, these findings add mechanistic insight into the association observed between PAI-1 and metabolic dysfunction pathologies which can accelerate cardiovascular aging.
Renal function and Klotho link
In addition to metabolic impacts, PAI-1 influences renal function, intersecting with aging pathways like the FGF23-Klotho axis. The kidneys regulate blood pressure and vascular tone through the renin-angiotensin-aldosterone system[85]. Impaired renal function can cause fluid retention and hypertension, increasing cardiac workload and contributing to heart failure. Chronic kidney disease (CKD) disrupts normal renin-angiotensin-aldosterone processes, leading to hypertension and an increased risk of CVD such as atherosclerosis and left ventricular hypertrophy.
The FGF23-Klotho axis is important for kidney function and mineral homeostasis[86]. FGF23, secreted by osteocytes, regulates phosphate levels by inhibiting renal phosphate reabsorption and suppressing active vitamin D synthesis. Klotho acts as a co-receptor for FGF23, facilitating its interaction with fibroblast growth factor receptor 1 (FGFR1) in the kidney. Klotho hypomorphs result from a spontaneous insertional mutation that disrupts the promoter region of the mouse klotho gene, reducing its expression without completely abolishing it, thus leading to a hypomorphic allele[87]. Although a complete knockout model of klotho exists, most studies use the mutant hypomorph (klotho-/-), which is accepted as a Klotho-deficient mouse[86]. In CKD, Klotho deficiency contributes to FGF23 resistance, exacerbating phosphate retention and promoting vascular calcification[88]. Klotho-deficient mice exhibit premature aging with a median lifespan of 61 days[87,89,90]. These klotho-/- mice have normal development, indicating Klotho's role in anti-aging rather than development. Our group showed that reducing circulating PAI-1 levels in klotho-/- mice rescues the premature aging phenotype[8]. We demonstrated that t-PA and u-PA cleave FGF23; thus, excess PAI-1 increases FGF23 levels by inhibiting t-PA and u-PA. Genetic deficiency and pharmacological inhibition of PAI-1 reduced circulating FGF23 levels. These results suggest that reducing PAI-1 levels may benefit renal function and overall aging.
Nuclear PAI-1
Emerging evidence suggests that PAI-1 may have roles within the nucleus. Gehlot et al. reported that PAI-1 localizes in the nucleus of human umbilical vein endothelial cells, using immunofluorescence and nuclear fractionation, and identified putative nuclear export signals[91]. Additionally, PAI-1 may translocate to the nucleus in bladder cancer, suggesting a possible role in transcriptional regulation[92]. In the context of colon cancer, it has been shown that PAI-1 directly binds to p65 of the NFκB complex and promotes the gene expression of NFκB’s transcriptional profile[93]. However, the characterization of a possible role for PAI-1 in the nucleus is still in its infancy and further experiments could elucidate potential DNA binding sites and mechanisms in the context of cardiovascular aging.
Evolutionary advantage of heterozygous PAI-1 deficiency in humans
Complementing experimental data, human studies of PAI-1 deficiency provide clinical insights into its impact on longevity and cardiovascular health. The evidence described above for PAI-1 as a therapeutic target for aging comes from animal and in vitro models. For translation to clinical use, compelling data from a “natural experiment” in a human population demonstrates the beneficial effects of lifelong PAI-1 reduction. In 1881, a male with a SERPINE1 genetic mutation married into a Swiss Amish community. Over a hundred years later, in 1992, descendants of this individual were identified as harboring a rare loss-of-function mutation in SERPINE1 (c.699_700dupTA)[94]. This mutation consists of a dinucleotide duplication leading to a frameshift in the mRNA and a truncated protein, and heterozygous carriers have ~50% of normal circulating PAI-1 levels[95]. This geographically isolated Amish community offers a rare real-time example of human evolution, where infants are effectively “randomized” as either carriers or non-carriers of the SERPINE1 mutation, enabling researchers to study the long-term effects of reduced PAI-1 levels.
We studied 177 members of this community, including 43 carriers of the mutation, and found that heterozygous carriers exhibited significantly longer leukocyte telomere length, lower fasting insulin levels, and protection from diabetes mellitus[95]. Further, carriers of the null SERPINE1 allele demonstrated preserved cardiovascular fitness and increased longevity by a median of 10 years. These findings suggest that the mutation may confer biological advantages in terms of metabolism, fertility, and protection from aging-related morbidities. The broader implications of this study extend beyond cardiovascular health, as age-related multisystem loss of resilience is closely linked to chronic diseases like metabolic disorders, cancer, and Alzheimer’s. Indeed, the Amish genetic profiles highlight PAI-1 as a mechanistic contributor to aging-related morbidity in humans, supporting the notion that its inhibition could serve as a powerful intervention for promoting human longevity.
CONCLUSION
High levels of PAI-1 are intricately associated with a spectrum of negative aging effects that underpin cardiovascular disease, including vascular stiffness, cellular senescence, immune evasion, metabolic dysfunction, and renal impairment. This review has described PAI-1’s role in these processes: it promotes vascular stiffness through collagen deposition and reduced nitric oxide bioavailability, amplifies senescence via senescence-associated secretory phenotype (SASP) factors and IGFBP-3 signaling, and facilitates immune evasion by upregulating PD-L1, thereby linking inflammaging to cardiovascular pathology [Figure 3]. Beyond the vasculature, PAI-1 exacerbates dyslipidemia by enhancing VLDL assembly and impairing fibrinolysis, contributes to renal dysfunction via the FGF23-Klotho axis, and may even influence nuclear transcriptional regulation, as emerging evidence suggests. Conversely, reducing PAI-1 levels through genetic deficiency or pharmacological inhibition mitigates these age-related conditions, as demonstrated in preclinical models and the remarkable longevity observed in humans with heterozygous PAI-1 deficiency.

Figure 3. PAI-1 drives cardiovascular aging through interconnected mechanisms: promoting vascular stiffness, amplifying senescence, and enabling immune evasion.
New technologies promise to deepen our understanding: single-cell RNA sequencing can reveal cell-specific PAI-1 expression patterns in aging vasculature, while advanced imaging techniques like atomic force microscopy offer novel insights into its mechanical effects on ECM. Additionally, CRISPR-based gene editing and high-throughput screening of PAI-1 inhibitors could accelerate the development of targeted therapies, potentially combined with senolytics or senomorphics for synergistic effects.
Given PAI-1’s central role in aging, significant efforts have been made to develop therapeutic strategies aimed at modulating its expression and function, including small molecules and antibody-based inhibitors; many of these approaches have been reviewed extensively elsewhere[6]. Several small molecule inhibitors have been developed to target PAI-1 activity, with preclinical and clinical data supporting their therapeutic potential. TM5441 and TM5614, selective PAI-1 inhibitors, have been shown to extend lifespan of klotho-/- mice[89] and mitigate senescence-associated fibrosis in the aorta of mice[38]. By inhibiting PAI-1’s function, these compounds not only restore fibrinolytic balance but may also influence senescence pathways, positioning them as potential geroprotective agents. Despite the advancements of inhibitors, the pleiotropic functions of PAI-1 necessitate a careful balance between therapeutic benefits and potential off-target effects. Additionally, long-term safety data are needed to establish their suitability for chronic administration. Future directions should focus on refining drug specificity and identifying patient populations most likely to benefit from PAI-1-targeted interventions. Combining PAI-1 inhibitors with other senolytic or senomorphic therapies may offer a comprehensive approach to combating age-related diseases and enhancing human healthspan.
DECLARATIONS
Authors’ contributions
Writing and generating figures: Khoddam A
Writing and editing: Vaughan D, Wilsbacher L
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by grants from the National Institute of Health to Vaughan D (R35HL171553) and from the American Heart Association to Khoddam A (25PRE1356859).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Cardiovascular diseases (CVDs). World Health Organization. 2021. Available from: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) [Last accessed on 22 Apr 2025].
2. Ageing and health. World Health Organization. 2024. Available from: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health [Last accessed on 22 Apr 2025].
3. Austad SN. The geroscience hypothesis: is it possible to change the rate of aging? In: Sierra F, Kohanski R, editors. Advances in geroscience. Cham: Springer International Publishing; 2016. pp. 1-36.
4. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186:243-78.
5. Abdellatif M, Rainer PP, Sedej S, Kroemer G. Hallmarks of cardiovascular ageing. Nat Rev Cardiol. 2023;20:754-77.
6. Sillen M, Declerck PJ. Targeting PAI-1 in cardiovascular disease: structural insights into PAI-1 functionality and inhibition. Front Cardiovasc Med. 2020;7:622473.
7. Loskutoff DJ, van Mourik JA, Erickson LA, Lawrence D. Detection of an unusually stable fibrinolytic inhibitor produced by bovine endothelial cells. Proc Natl Acad Sci USA. 1983;80:2956-60.
8. Eren M, Place AT, Thomas PM, Flevaris P, Miyata T, Vaughan DE. PAI-1 is a critical regulator of FGF23 homeostasis. Sci Adv. 2017;3:e1603259.
9. Gerenu G, Martisova E, Ferrero H, et al. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer's neuropathology and cognitive deficits. Biochim Biophys Acta Mol Basis Dis. 2017;1863:991-1001.
10. Cai D, Holm JM, Duignan IJ, et al. BDNF-mediated enhancement of inflammation and injury in the aging heart. Physiol Genomics. 2006;24:191-7.
11. Elzi DJ, Lai Y, Song M, Hakala K, Weintraub ST, Shiio Y. Plasminogen activator inhibitor 1-insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc Natl Acad Sci USA. 2012;109:12052-7.
12. Wang H, Zhang Y, Heuckeroth RO. PAI-1 deficiency reduces liver fibrosis after bile duct ligation in mice through activation of tPA. FEBS Lett. 2007;581:3098-104.
13. Morrow GB, Whyte CS, Mutch NJ. A serpin with a finger in many PAIs: PAI-1’s central function in thromboinflammation and cardiovascular disease. Front Cardiovasc Med. 2021;8:653655.
14. Tjärnlund-Wolf A, Brogren H, Lo EH, Wang X. Plasminogen activator inhibitor-1 and thrombotic cerebrovascular diseases. Stroke. 2012;43:2833-9.
15. Rizzo MR, Ragno E, Barbieri M, et al. Elevated plasma activator inhibitor 1 is not related to insulin resistance and to gene polymorphism in healthy centenarians. Atherosclerosis. 2002;160:385-90.
16. Samad F, Loskutoff DJ. Tissue distribution and regulation of plasminogen activator inhibitor-1 in obese mice. Mol Med. 1996;2:568-82.
17. Eren M, Painter CA, Atkinson JB, Declerck PJ, Vaughan DE. Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation. 2002;106:491-6.
18. Eren M, Gleaves LA, Atkinson JB, King LE, Declerck PJ, Vaughan DE. Reactive site-dependent phenotypic alterations in plasminogen activator inhibitor-1 transgenic mice. J Thromb Haemost. 2007;5:1500-8.
19. Yamamoto K, Takeshita K, Kojima T, Takamatsu J, Saito H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res. 2005;66:276-85.
20. Laurent S, Boutouyrie P, Asmar R, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37:1236-41.
21. Regnault V, Lacolley P, Laurent S. Arterial stiffness: from basic primers to integrative physiology. Annu Rev Physiol. 2024;86:99-121.
22. Lyle AN, Raaz U. Killing me unsoftly: causes and mechanisms of arterial stiffness. Arterioscler Thromb Vasc Biol. 2017;37:e1-11.
23. Hickson SS, Butlin M, Graves M, et al. The relationship of age with regional aortic stiffness and diameter. JACC Cardiovasc Imaging. 2010;3:1247-55.
24. Kawasaki T, Sasayama S, Yagi S, Asakawa T, Hirai T. Non-invasive assessment of the age related changes in stiffness of major branches of the human arteries. Cardiovasc Res. 1987;21:678-87.
25. Vatner SF, Zhang J, Vyzas C, Mishra K, Graham RM, Vatner DE. Vascular stiffness in aging and disease. Front Physiol. 2021;12:762437.
26. Chu CQ, Quan T. Fibroblast Yap/Taz signaling in extracellular matrix homeostasis and tissue fibrosis. J Clin Med. 2024;13:3358.
27. Ribeiro-Silva JC, Nolasco P, Krieger JE, Miyakawa AA. Dynamic crosstalk between vascular smooth muscle cells and the aged extracellular matrix. Int J Mol Sci. 2021;22:10175.
28. Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol. 2013;229:298-309.
29. Kennelly PJ, Botham KM, McGuinness OP, Rodwell VW, Weil PA. Harper’s illustrated biochemistry, 32nd edition. 2023. Available from: https://archive.org/details/harpers-illustrated-biochemistry-thirty-second-edition-2023/mode/2up [Last accessed on 22 Apr 2025].
30. Kolpakov V, Gordon D, Kulik TJ. Nitric oxide-generating compounds inhibit total protein and collagen synthesis in cultured vascular smooth muscle cells. Circ Res. 1995;76:305-9.
31. Diebold I, Kraicun D, Bonello S, Görlach A. The ‘PAI-1 paradox’ in vascular remodelling. Thromb Haemost. 2008;100:984-91.
32. Zaman AK, French CJ, Schneider DJ, Sobel BE. A profibrotic effect of plasminogen activator inhibitor type-1 (PAI-1) in the heart. Exp Biol Med. 2009;234:246-54.
33. Lackie PM. Molecular portfolios: cells interacting with matrix in repairing airway epithelium: this editorial discusses the findings of the paper in this issue by Stevens et al. [3] pp. 1901-10. Clin Exp Allergy. 2008;38:1840-3.
34. Gramley F, Lorenzen J, Plisiene J, et al. Decreased plasminogen activator inhibitor and tissue metalloproteinase inhibitor expression may promote increased metalloproteinase activity with increasing duration of human atrial fibrillation. J Cardiovasc Electrophysiol. 2007;18:1076-82.
36. Kortlever RM, Nijwening JH, Bernards R. Transforming growth factor-beta requires its target plasminogen activator inhibitor-1 for cytostatic activity. J Biol Chem. 2008;283:24308-13.
37. Pedroja BS, Kang LE, Imas AO, Carmeliet P, Bernstein AM. Plasminogen activator inhibitor-1 regulates integrin αvβ3 expression and autocrine transforming growth factor β signaling. J Biol Chem. 2009;284:20708-17.
38. Boe AE, Eren M, Murphy SB, et al. Plasminogen activator inhibitor-1 antagonist TM5441 attenuates Nω-nitro-L-arginine methyl ester-induced hypertension and vascular senescence. Circulation. 2013;128:2318-24.
39. Garcia V, Park EJ, Siragusa M, et al. Unbiased proteomics identifies plasminogen activator inhibitor-1 as a negative regulator of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2020;117:9497-507.
40. Khoukaz HB, Vadali M, Schoenherr A, et al. PAI-1 regulates the cytoskeleton and intrinsic stiffness of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2024;44:2191-203.
41. Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao PS. eNOS activity is reduced in senescent human endothelial cells: preservation by hTERT immortalization. Circ Res. 2001;89:793-8.
42. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729-40.
43. Han Y, Kim SY. Endothelial senescence in vascular diseases: current understanding and future opportunities in senotherapeutics. Exp Mol Med. 2023;55:1-12.
44. Bloom SI, Islam MT, Lesniewski LA, Donato AJ. Mechanisms and consequences of endothelial cell senescence. Nat Rev Cardiol. 2023;20:38-51.
45. Stefens SJM, van Vliet N, IJpma A, et al. Increased vascular smooth muscle cell senescence in aneurysmal Fibulin-4 mutant mice. NPJ Aging. 2024;10:31.
46. Gasek NS, Kuchel GA, Kirkland JL, Xu M. Strategies for targeting senescent cells in human disease. Nat Aging. 2021;1:870-9.
47. Burton DG, Matsubara H, Ikeda K. Pathophysiology of vascular calcification: pivotal role of cellular senescence in vascular smooth muscle cells. Exp Gerontol. 2010;45:819-24.
48. Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232-6.
49. Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184-9.
50. Zhang X, Pearsall VM, Carver CM, et al. Rejuvenation of the aged brain immune cell landscape in mice through p16-positive senescent cell clearance. Nat Commun. 2022;13:5671.
51. Zhu Y, Tchkonia T, Pirtskhalava T, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14:644-58.
52. Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. 2017;21:21-8.
53. Tao W, Yu Z, Han JJ. Single-cell senescence identification reveals senescence heterogeneity, trajectory, and modulators. Cell Metab. 2024;36:1126-43.e5.
54. Vaughan DE, Rai R, Khan SS, Eren M, Ghosh AK. Plasminogen activator inhibitor-1 is a marker and a mediator of senescence. Arterioscler Thromb Vasc Biol. 2017;37:1446-52.
55. Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877-84.
56. Cesari M, Pahor M, Incalzi RA. Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc Ther. 2010;28:e72-91.
57. Eren M, Boe AE, Klyachko EA, Vaughan DE. Role of plasminogen activator inhibitor-1 in senescence and aging. Semin Thromb Hemost. 2014;40:645-51.
58. Sillen M, Declerck PJ. A narrative review on plasminogen activator inhibitor-1 and its (patho)physiological role: to target or not to target?. Int J Mol Sci. 2021;22:2721.
59. Sun T, Ghosh AK, Eren M, Miyata T, Vaughan DE. PAI-1 contributes to homocysteine-induced cellular senescence. Cell Signal. 2019;64:109394.
60. Ghosh AK, Rai R, Park KE, et al. A small molecule inhibitor of PAI-1 protects against doxorubicin-induced cellular senescence. Oncotarget. 2016;7:72443-57.
61. Uryga AK, Grootaert MOJ, Garrido AM, et al. Telomere damage promotes vascular smooth muscle cell senescence and immune cell recruitment after vessel injury. Commun Biol. 2021;4:611.
62. Saggau C, Bacher P, Esser D, et al. Autoantigen-specific CD4+ T cells acquire an exhausted phenotype and persist in human antigen-specific autoimmune diseases. Immunity. 2024;57:2416-32.e8.
63. Onorati A, Havas AP, Lin B, et al. Upregulation of PD-L1 in senescence and aging. Mol Cell Biol. 2022;42:e0017122.
64. Cho HJ, Hwang JA, Yang EJ, et al. Nintedanib induces senolytic effect via STAT3 inhibition. Cell Death Dis. 2022;13:760.
65. Xu M, Palmer AK, Ding H, et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife. 2015;4:e12997.
66. Xu M, Tchkonia T, Ding H, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci USA. 2015;112:E6301-10.
67. Wang TW, Johmura Y, Suzuki N, et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature. 2022;611:358-64.
68. Suero-Abreu GA, Zanni MV, Neilan TG. Atherosclerosis with immune checkpoint inhibitor therapy: evidence, diagnosis, and management: JACC: CardioOncology state-of-the-art review. JACC CardioOncol. 2022;4:598-615.
69. Ibrahim AA, Fujimura T, Uno T, et al. Plasminogen activator inhibitor-1 promotes immune evasion in tumors by facilitating the expression of programmed cell death-ligand 1. Front Immunol. 2024;15:1365894.
70. Lin LL, Nayak B, Osmulski PA, et al. PAI-1 uncouples integrin-β1 from restrain by membrane-bound β-catenin to promote collagen fibril remodeling in obesity-related neoplasms. Cell Rep. 2024;43:114527.
71. Hendrikson J, Liu Y, Ng WH, et al. Ligand-mediated PAI-1 inhibition in a mouse model of peritoneal carcinomatosis. Cell Rep Med. 2022;3:100526.
72. Kortlever RM, Bernards R. Senescence, wound healing and cancer: the PAI-1 connection. Cell Cycle. 2006;5:2697-703.
73. Chibaya L, Snyder J, Ruscetti M. Senescence and the tumor-immune landscape: Implications for cancer immunotherapy. Semin Cancer Biol. 2022;86:827-45.
74. Lin LL, Kost ER, Lin CL, et al. PAI-1-dependent inactivation of SMAD4-modulated junction and adhesion complex in obese endometrial cancer. Cell Rep. 2020;33:108253.
75. Kubala MH, DeClerck YA. The plasminogen activator inhibitor-1 paradox in cancer: a mechanistic understanding. Cancer Metastasis Rev. 2019;38:483-92.
76. Bastard J, Piéroni L, Hainque B. Relationship between plasma plasminogen activator inhibitor 1 and insulin resistance. Diabetes Metab Res Rev. 2000;16:192-201.
77. Nordt TK, Sawa H, Fujii S, Sobel BE. Induction of plasminogen activator inhibitor type-1 (PAI-1) by proinsulin and insulin in vivo. Circulation. 1995;91:764-70.
78. Dai W, Zhang H, Lund H, et al. Intracellular tPA-PAI-1 interaction determines VLDL assembly in hepatocytes. Science. 2023;381:eadh5207.
79. Levine JA, Oleaga C, Eren M, et al. Role of PAI-1 in hepatic steatosis and dyslipidemia. Sci Rep. 2021;11:430.
80. Levine J, Sun T, Eren M, et al. OR22-6 reversal of diet induced metabolic syndrome in mice with an orally active small molecule inhibitor of PAI-1. J Endocr Soc. 2019;3:OR22-6.
81. Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res. 2018;123:825-48.
82. Folsom AR, Qamhieh HT, Wing RR, et al. Impact of weight loss on plasminogen activator inhibitor (PAI-1), factor VII, and other hemostatic factors in moderately overweight adults. Arterioscler Thromb. 1993;13:162-9.
83. Bareja A, Lee DE, Ho T, et al. Liver-derived plasminogen mediates muscle stem cell expansion during caloric restriction through the plasminogen receptor Plg-R(KT). Cell Rep. 2024;43:113881.
84. Hong SH, Choi KM. Sarcopenic obesity, insulin resistance, and their implications in cardiovascular and metabolic consequences. Int J Mol Sci. 2020;21:494.
85. Silverthorn DU. Human physiology: an integrated approach. 2020. Available from: https://archive.org/details/humanphysiologyi0000silv_x2f9 [Last accessed on 22 Apr 2025].
86. Buchanan S, Combet E, Stenvinkel P, Shiels PG. Klotho, aging, and the failing kidney. Front Endocrinol. 2020;11:560.
87. Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45-51.
88. Hu MC, Shi M, Zhang J, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22:124-36.
89. Eren M, Boe AE, Murphy SB, et al. PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc Natl Acad Sci USA. 2014;111:7090-5.
91. Gehlot P, Brünnert D, Kaushik V, et al. Unconventional localization of PAI-1 in PML bodies: a possible link with cellular growth of endothelial cells. Biochem Biophys Rep. 2024;39:101793.
92. Furuya H, Sasaki Y, Chen R, et al. PAI-1 is a potential transcriptional silencer that supports bladder cancer cell activity. Sci Rep. 2022;12:12186.
93. Zhang D, Zhang JW, Xu H, et al. Therapy-induced senescent tumor cell-derived extracellular vesicles promote colorectal cancer progression through SERPINE1-mediated NF-κB p65 nuclear translocation. Mol Cancer. 2024;23:70.
94. Fay WP, Parker AC, Condrey LR, Shapiro AD. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. 1997;90:204-8.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].