

Coke deposition mechanisms of propane dehydrogenation on different sites of Al2O3 supported PtSn catalysts

Abstract
Propane dehydrogenation (PDH) Pt-based catalysts are facing the serious challenge of coke deactivation. The locations would greatly influence the coke formation, while the detailed mechanism is not fully explored. Herein, the coke mechanisms on different locations including Al2O3, Sn, Pt, and Pt-Sn sites were deeply investigated via in situ Fourier transform infrared spectroscopy (FTIR) technology, and the key factors triggering catalyst coke deactivation were proposed. Excessive dehydrogenation of propyl species is a crucial initial step in the formation of coke, whether at metal sites or supports. These propyl species on Al2O3 supports then cyclize to form monocyclic aromatic and bicyclic aromatic species, while those on SnOx sites cyclize to form monocyclic aromatic species. As for the Al2O3 supported PtSn catalysts, the strong dehydrogenation function and the interaction between Pt and

Keywords
INTRODUCTION
Pt-based catalysts exhibit excellent catalytic activity in the direct dehydrogenation of propane to propylene, while they face a severe challenge of coke deactivation[1-4]. Frequent regeneration not only involves complex processes and huge energy consumption but also cannot fundamentally resolve the problem of coke deactivation[2,5]. Consequently, researchers have developed extensive optimized strategies from various perspectives to suppress coke deactivation, including constructing active phases, selecting carriers, enhancing interactions between metal and carriers, and controlling reaction pathways[6-10]. Among these, adding tin as a promoter to modify, both electronically and geometrically, the structure of the supported Pt on Al2O3 carriers is an effective approach[6,7]. Although this can alleviate the coke deposition problem to some extent, a large amount of coke still forms on the Al2O3 supported PtSn catalyst. A fundamental resolution to the coke deposition necessitates a deep understanding of the key initiation steps and the dynamic evolution mechanism of coke species in propane dehydrogenation (PDH).
The processes of coke formation are complicated, involving complex surface reactions and various intermediates in the PDH process[11,12]. The specific effect brought by coke accumulation significantly depends on the catalytic system and the location of coke deposition. In Pt-based catalysts, the coke positions have been studied, which are classified into three locations, i.e., metallic Pt, the boundary between the Pt and the support, and the support[13,14]. Researchers have associated different site characteristics with their propensity to cause coke deposition and discovered significant variations in the structure and properties of coke formed at different reaction sites[13].
Coke deposition initiated at metallic Pt active sites mainly originates from their strong dehydrogenation function. According to the current mechanism for the PDH reaction, propane molecules adsorb onto the metal sites and undergo two consecutive dehydrogenations, forming a propylene precursor that strongly adsorbs on the metal sites and undergoes excessive dehydrogenation, leading to the formation of coke[8,15]. Numerous studies indicate that larger Pt particles are prone to coke deposition, compared with the small-sized Pt clusters[16,17]. This is mainly attributed to the strong adsorption of propylene precursors on large-sized Pt particles, resulting in excessive dehydrogenation. Besides, the coordination structure of the Pt active phase can also affect coke formation. The coordination-saturated Pt active sites facilitate side reactions, such as C–C cleavage and deep dehydrogenation, thereby forming coke deposits[6,18].
The carrier is another crucial factor in the initiation of coke deposition[19]. Alumina, used as the carrier in industrial Pt-based PDH catalysts, has pronounced acidic characteristics that enable it to catalyze a series of side reactions, such as oligomerization, cyclization, and condensation of the desorbed product olefins, leading to the formation of coke with a high hydrogen-to-carbon ratio[20]. Numerous studies have shown that reducing the acidity of the carrier can significantly decrease the occurrence of cracking and polymerization reactions, enhance the selectivity for propylene in the modified samples, and markedly inhibit the coke deactivation of catalysts[21]. However, some investigators also proposed that when the acidity of the catalyst is too low, the coke amount increases rather than decreases[13,14]. They attributed it to the synergistic effect between metal and acidic sites that inhibited the formation of coke deposition. Therefore, it is evident that the formation of coke deposition during PDH is influenced by the carrier acidity and the synergistic interaction between the carrier and metal sites.
In addition to metal sites and carriers, the reaction situation is a key factor affecting coke deposition behavior in PDH[8,12,22]. In practical propylene production, hydrogen as a co-feeding gas has been utilized to inhibit the deactivation rate, obviously improving the stability of Pt-based catalysts[23]. This inhibitory mechanism is usually attributed to two main factors: (i) the desorption barrier of propylene can be reduced; (ii) metallic active sites can be preserved with the existence of hydrogen[24]. However, the introduction of hydrogen has a large influence on the properties of metal sites and the support acidity, inevitably leading to different coke deposition behaviors.
Hydrogen dissociates at metal sites and forms metal-hydride species (M-H), which possess different adsorption capabilities for propylene precursors from the metallic active phase, thereby significantly influencing coke deposition[25-27]. The hydrogen spillover caused by the metallic Pt sites, forming active hydrogen species, also significantly impacts coke formation[28,29]. Besides, the spillover of these active hydrogen species onto the carrier can alter its surface properties[30,31], triggering another crucial factor of coke formation. Obviously, different sites and reaction situations substantially influence coke formation in PDH with a high degree of interconnectivity between them. Due to the complex interactions of the various factors and the complexity of the reaction mechanisms, the coke formation mechanisms at different sites in PDH are not yet well understood which evidently limits the progress in research on inhibiting coke deactivation. Revealing the coke formation mechanisms in the Pt-based catalyst system could significantly advance the development of de-coking technologies in the PDH field.
Herein, the Al2O3 supported PtSn catalyst, which is widely used and has excellent performance in PDH, is used as the research object. Starting with the key initiation steps of coke deposition in PDH, the formation mechanisms of coke at different locations are deeply analyzed using a Fourier transform infrared spectroscopy (FTIR) method, and an active site structure that does not undergo coke deactivation is proposed. More specifically, the relations between the location of coke, structures of active phase and coke deactivation mechanism are established. The FTIR spectra can provide very important information on the amount and nature of coke and on its effect on the catalyst acidity.
EXPERIMENTAL
Catalyst preparation
Al2O3 (99%, Sasol Germany GmbH) and Sn-Al2O3 (99%, Sasol Germany GmbH) were calcined at 650 °C for 4 h to make γ-Al2O3 support and Sn-γ-Al2O3, respectively. The obtained catalysts were named as the Al2O3 and Sn-Al2O3. The Sn content of the Sn-Al2O3 sample is 0.3 wt%.
The Pt/Al2O3 catalyst was synthesized by a wet impregnation method, in which the theoretical amount of Pt was 0.5 wt%. First, 13.25 mg of H2PtCl6·6H2O (99.5%, Sinopharm Chemical Reagent Co. Ltd.) were diluted in appropriate amounts of water. Subsequently, the solution containing the metal precursors was impregnated into 1.0 g of the above calcined Al2O3. After impregnation, the samples were stirred for 15 min, maintained at room temperature for 6 h, and then dried at 50 °C for 12 h. This was followed by calcination in 20% O2/N2 at 600 °C for 2 h. Sample reduction was conducted in pure H2 atmosphere at
Catalyst characterization
The crystalline structures of the samples were confirmed using a Bruker D8 Advance X-ray diffraction (XRD) instrument. This analysis was conducted using Cu Kα radiation, characterized by a wavelength of 0.154 nm, under an operating voltage of 3 kV. A transmission electron microscopy (TEM) image was recorded on a JEOL-2100F microscope, which was operated at an accelerating voltage of 200 kV. The analysis utilizing X-ray photoelectron spectroscopy (XPS) was conducted employing an ESCALAB Xi+ instrument from Thermo Fisher Scientific, which utilized Al Kα radiation for the X-ray source. All samples underwent a reduction process at 500 °C for 1 h in an atmosphere of pure H2, after which they were hermetically sealed and stored in vials. The hydrogen temperature programmed reduction (H2-TPR) analysis was executed using a BSD-Chem C200 system. This involved flowing a 10% H2/Ar mixture (with a flow rate of 30 mL/min) over a 100 mg sample (with a particle size of 40-60 mesh), while heating at a rate of 10 °C/min per minute until reaching 700 °C. The hydrogen temperature programmed desorption (H2-TPD) analysis was conducted using a BSD-Chem C200 device. In a typical procedure, 50 mg of the catalyst was initially treated under a 10% H2/Ar flow (30 mL/min) at 500 °C for 2 h, followed by a 30-minute He purge at the same flow rate. To facilitate hydrogen absorption, a H2/Ar mixture was flowed over the catalyst for
FTIR spectra were recorded on a Thermo Scientific Nicolet iS50 FTIR spectrometer. All spectra were collected as an average of 64 scans at 4 cm-1 resolutions. The difference spectrum is obtained by subtracting the collected infrared (IR) spectra from the background spectra. (1) CO-adsorption experiment: the sample was initially reduced with H2/Ar (30 mL/min) for 1 h and subsequently cooled to 30 °C. Following this, it was exposed to a 10% CO/Ar mixture until equilibrium was reached. Then, the cell was purged with Ar flow to remove the physically adsorbed CO, with IR spectra being gathered continuously until stabilization; (2) Pyridine-adsorption experiment: the samples were activated at 400 °C for 2 h under a vacuum setup
Catalytic tests
The PDH performance was conducted in a tubular quartz reactor with a fixed bed under atmospheric pressure. The reactor’s temperature was regulated using a Golden Eagle temperature controller (Series 708P). In each test, 0.1 g of the catalyst was utilized. A gaseous mixture containing C3H8 and H2 in a 1:1 volume ratio was introduced over the catalyst layer at 600 °C. Product analysis was performed using an inline gas chromatograph. For the separation and detection of H2, C3H8, C3H6, C2H4, C2H6, CH4, and C4-C6 in the effluent, KB-1 (8 m × 0.32 mm × 0.50 μm) and TDX-01 (3 mm × 1 m) columns were employed. The conversion of propane and the selectivity for the various products were determined by:
Propane conversion:
Selectivity of products:
where [C3H8]in, [C3H8]out and [C3H6]out were the imported molar concentration of C3H8, the outlet molar concentration of C3H8 and C3H6 in the effluent gas, respectively.
RESULTS AND DISCUSSION
Evolution path of coke precursors on different sites of Al2O3 supported PtSn catalysts
The coke formation on Al2O3 supported PtSn catalysts primarily occurs at metal sites, metal-support interface and supports, with distinct mechanisms at each location. However, the current research on the coke deposition mechanism at different sites and the interactive effects between various sites remain unclear [Supplementary Table 1]. To investigate the coke formation mechanisms at these sites, four model catalysts, Al2O3, Sn/Al2O3, Pt/Al2O3, and Pt/Sn-Al2O3, were designed and prepared. All samples maintain the typical crystal phase structure of γ-Al2O3 [Supplementary Figure 1] but have different active components. These four catalysts exhibit similar crystalline morphology and size [Supplementary Figures 2 and 3].
To investigate the formation mechanism of coke precursors on catalysts, a hyphenated technique combining pulse, high temperature and in situ IR spectroscopy was utilized to analyze the types and formation mechanisms of these precursors [Figure 1]. The pulse experiments were carried out at 600 °C by pulses of pure propane (pulse volume = 0.50 cm3 STP) to the in-situ cell, which was maintained under Ar flow (30 mL/min). After the pulse, FTIR spectra were continuously recorded for five minutes. Table 1 exhibits the summary of the vibrational modes and positions of adsorption peaks. When 0.5 mL of C3H8 is pulsed into the in situ cell at 600 °C, strong absorption peaks at 2,872-2,985 cm-1 attributed to the stretching vibrations of methyl C–H bonds in C3H8 are observed [Figure 1][32,33]. These four catalysts show distinct formation processes of coke precursor due to differences in their active components.
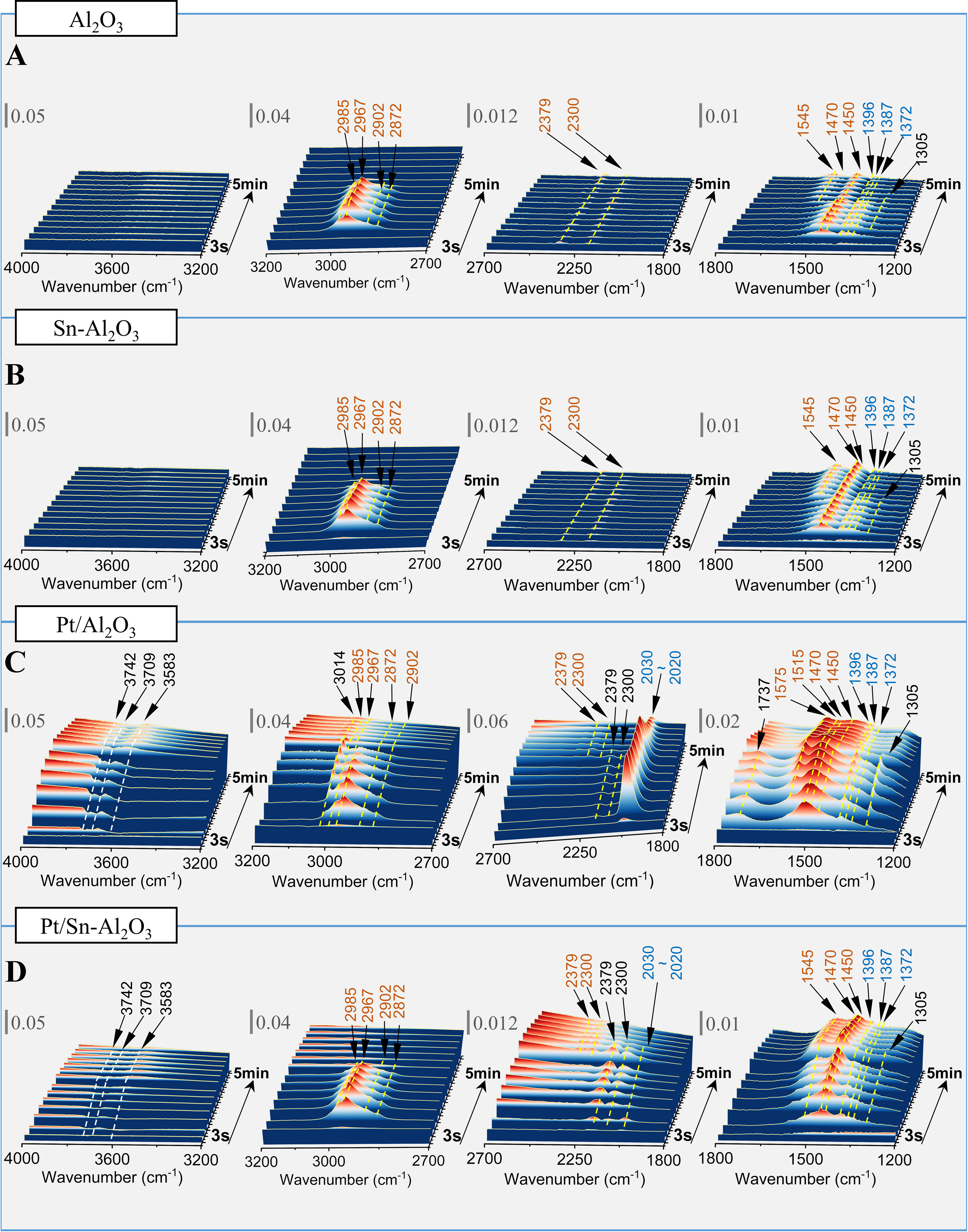
Figure 1. The coke mechanism of PDH on various locations. FTIR differential spectra following propane pulse adsorption for (A) Al2O3, (B) Sn-Al2O3, (C) Pt/Al2O3 and (D) Pt/Sn-Al2O3. The pulse experiments were carried out at 600 °C by pulses of pure propane (pulse volume = 0.50 cm3 STP) to the in situ cell at Ar (30 mL/min) atmosphere. The yellow dashed line represents the adsorption peak, while the white dashed line represents the inverted peak caused by the occupation of adsorption sites. PDH: Propane dehydrogenation; FTIR: Fourier transform infrared spectroscopy; STP: standard temperature and pressure.
Summary of the vibrational modes and positions of adsorption peaks (1,200-4,000 cm-1)
| Species | Vibrational mode | Band position (cm-1) | |
| This work | Literature range | ||
| Olefin | δm (C–H) | 1,305 | 1,295-1,310[34] |
| 1-Propylidyne | δs (C–H) | 1,372, 1,387 | 1,370-1,377[34,35] |
| 1-Propylidene 2-Propylidene | δs (C–H) | 1,396 | 1,392-1,398[34,35] |
| 1-Propyl 2-Propyl | δs (C–H) | 1,450 | 1,450-1,458[34,35] |
| Benzene | ν (C=C) | 1,470 | 1,450-1,650[43-46] |
| Naphthalene | ν (C=C) | 1,545 | 1,488-1,545[43-46] |
| Polyaromatic | ν (C=C) | 1,575 | 1,575-1,602[43-46] |
| Acetone | δs (C–H) ν (C=O) δ (C–H) | 3,014 1,737 1,440 | 3,000-3,300[40] 1,668-1,725[40-42] 1,434-1,436[40-42] |
| Alkyne | ν (C≡C) | 2,104-2,190 | 2,100-2,260[43] |
| Propane Al2O3 | νvs (C=C) | 2,985-2,872 | 2,875-2,990[44] |
| νvs (H–O) | 3,700-3,200 | 3,200-3,700[45] | |
For the Al2O3 sample, four peaks between 1,372 and 1,450 cm-1 attributed to the bending vibrations of C–H in CH2 and CH3 groups are observed at the initial stage of pulsing propane [Figure 1A]. Specifically, the peak at 1,450 cm-1 is associated with 1-propyl and 2-propyl vibrations, 1,396 cm-1 with 1-propylidene and
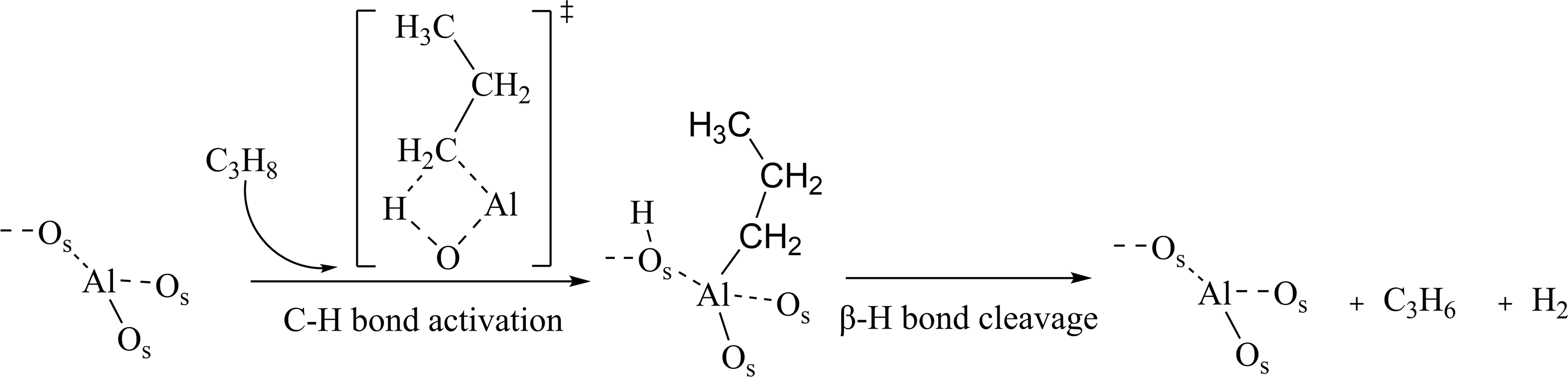
Scheme 1. Reaction mechanism for PDH on the Al2O3 sample. PDH: Propane dehydrogenation.
Furthermore, the FTIR spectrum of Al2O3 also exhibits absorption peaks at 1,470-1,600 cm-1 and 2,100-
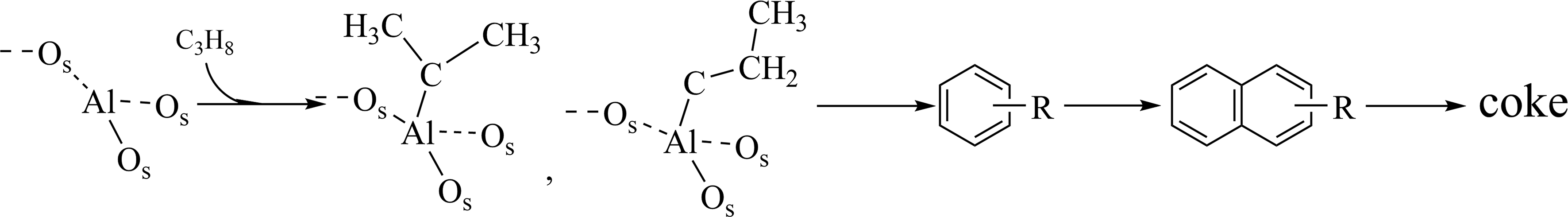
Scheme 2. Formation mechanism of coke precursors on the Al2O3 sample.
Doping Sn species into alumina alters its acidity. Similar to alumina, Sn-Al2O3 only contains Lewis acid sites, but both its acid amount and strength are significantly higher than those of Al2O3 [Supplementary Figures 4 and 5][49]. XPS results show that the Sn species in Sn-Al2O3 mainly exists in the form of Sn4+ [Supplementary Figure 8]. This is further confirmed by the absence of significant Sn species reduction peaks in H2-TPR of Sn-Al2O3 [Supplementary Figure 9][50]. These results indicate that the main Sn species in Sn-Al2O3 is SnOx. Therefore, the acidity change in the Sn-Al2O3 catalyst primarily originates from SnOx species, which show Lewis acidity. When pulsing propane into the Sn-Al2O3 catalyst, the absorption peaks at 1,396, 1,470, and 1,545 cm-1 intensify [Figure 1B]. The intensities of peaks at 1,470 and 1,545 cm-1 belonging to C=C stretching vibrations for monocyclic and polycyclic aromatic species in Sn-Al2O3 are notably higher than those in
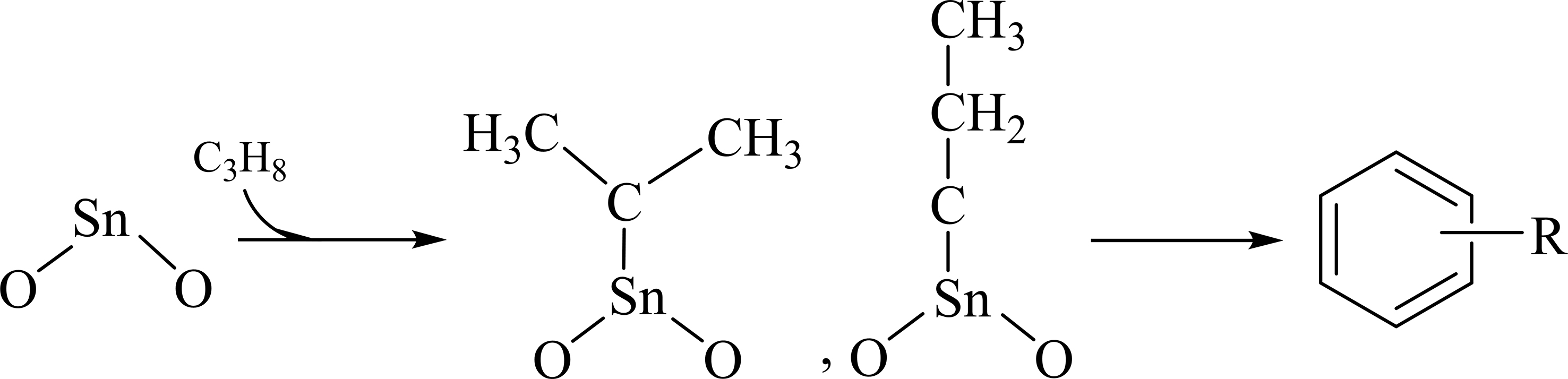
Scheme 3. Formation mechanism of coke precursors on the Sn-Al2O3 sample.
After loading Pt, its strong dehydrogenation capability significantly influences the coke deposition mechanism on the catalyst, warranting a discussion on the formation mechanism of coke precursors on Pt/Al2O3 [Figure 1C]. Compared to Al2O3 and Sn-Al2O3 catalysts [Figure 1A and B], the intensity of δs (C–H) absorption peaks for propylidene and propyl groups at 1,372-1,450 cm-1, and δm (C–H) absorption peaks for olefins at 1,305 cm-1 significantly increase. This indicates intense dehydrogenation reactions of C3H8 molecules at Pt sites with strong dehydrogenation ability, forming a large amount of propyl and olefin species. The intensity of peak at 2,104-2,190 cm-1 increases with the intensification of the peaks at 1,305-1,396 cm-1. Hence, further dehydrogenation of propylidyne can form adsorbed diene or acetylene species, whose further cyclization can produce monocyclic aromatics. Different from the Al2O3 and Sn-Al2O3 catalysts [Figure 1A and B], the Pt/Al2O3 catalyst rapidly forms a strong vibration peak at 1,515 cm-1 at the reaction onset, situated between the peak of polycyclic aromatics at 1,545 cm-1 and the peak of monocyclic aromatics at 1,470 cm-1. This peak is attributed to the cycloalkyl benzene coke precursors[52,53]. As the peak areas of monocyclic aromatics at 1,470 cm-1 and cycloalkyl benzene species at 1,515 cm-1 decrease, the polycyclic aromatic peak at 1,575 increases, suggesting that the polycyclic aromatic coke precursors are primarily derived from the condensation of monocyclic aromatic and cycloalkyl benzene species.
Interestingly, an absorption peak at ~1,737 cm-1 is observed in the Pt/Al2O3 [Figure 1C], which is attributed to the ν (C–O) vibration[54,55]. Concurrently, a significant negative peak appears at 3,600-3,800 cm-1, indicating that the hydroxyl groups in the alumina are occupied. These results suggest that the C in coke precursors is linked to O in Al2O3, forming C–O bonds. Airaksinen et al. and Finocchio et al. also reported the presence of acetone-like species in coke precursors on alumina, which further confirms the formation of the C–O bonds in our study[56,57]. Comparative analysis of the change in these C–O species absorption peaks during the reaction indicates that the concentration of C–O species initially increases rapidly with reaction time, then gradually decreases until it completely vanishes. Notably, the absorption peak of C–O species was only observed in the FTIR difference spectra of Pt/Al2O3 sample, not in Al2O3 or Sn-Al2O3
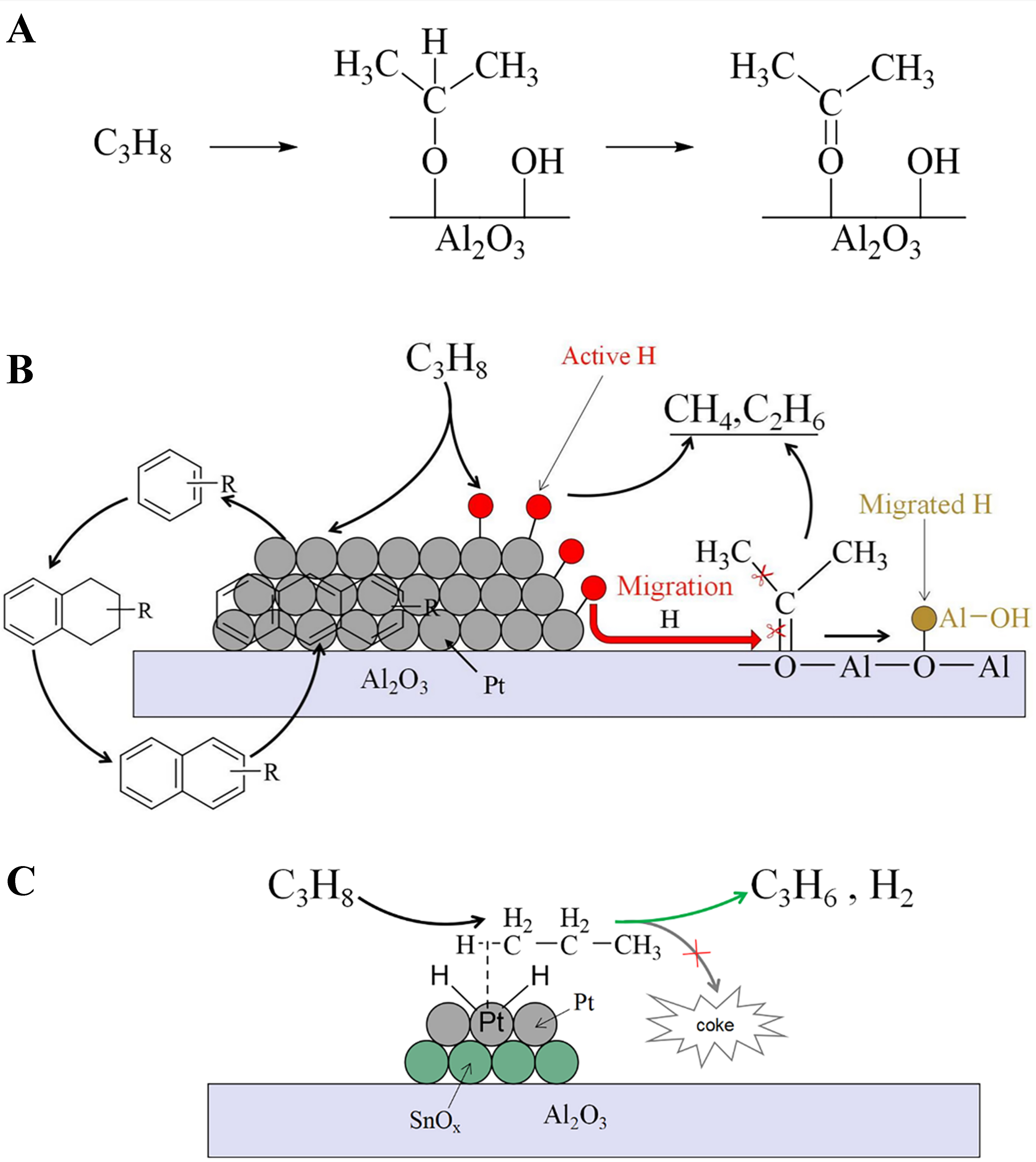
Scheme 4. (A) The deep dehydrogenation of propane at the oxygen species of Al2O3 support in Pt/Al2O3 sample; (B) Formation mechanism of the coke precursor and dehydrogenation mechanism on Pt/Al2O3 in PDH, the red ball and line represent the active hydrogen species; (C) Formation mechanism of the coke precursor and dehydrogenation mechanism on the Pt/Sn-Al2O3 catalyst. PDH: Propane dehydrogenation.
Another significant difference between Pt/Al2O3 [Figure 1C] and Al2O3 [Figure 1A] or Sn-Al2O3 [Figure 1B] is the strong absorption peak at 2,020-2,030 cm-1, which is attributed to Pt–H species or active hydrogen species produced by the activation of hydrogen on Pt sites[58]. The large amount of hydrogen spillover desorption peaks at 200-500 °C in H2-TPD for Pt/Al2O3 further confirms substantial active hydrogen species [Supplementary Figure 10]. There is a correlation between active hydrogen species and C–O species absorption peaks. The formation of active hydrogen species is accompanied by the appearance of C–O species. As the C–O species decrease, the content of active hydrogen species also diminishes, and the corresponding inverted peak at the surface hydroxyl groups becomes significantly smaller. This indicates that active hydrogen spills over from Pt sites to the support surface, reacting with C–O species to form hydrocarbon molecules, thereby restoring the alumina hydroxyl groups [Scheme 4B].
Based on scanning transmission electron microscopy (STEM) [Supplementary Figure 3] and CO adsorption IR spectroscopy [Supplementary Figure 11] results, the geometric effect of Sn leads to a smaller size of metal clusters in the Pt/Sn-Al2O3 catalyst (1.51 nm) compared to Pt/Al2O3 (1.74 nm). XPS and H2-TPR results confirm that Pt species in the Pt/Sn-Al2O3 catalyst exist in a lower coordination number and a partially oxidized state [Supplementary Figures 9 and 12, Supplementary Table 2][59-61]. Correspondingly, the main Sn species in the Pt/Sn-Al2O3 catalyst consist of Sn4+ (495.5, 487.0 eV) accounting for 74.0%, Sn2+ (494.6,
The 2,020-2,031 cm-1 region is attributed to the vibrational peaks resulting from the interaction between hydrogen and Pt. Compared to the Pt/Al2O3 catalyst [Figure 1C], the peak of Pt–H species in the Pt/Sn-
Key factors affecting coke deposition of Al2O3 supported PtSn catalysts
Structures of Pt active phase
The coke formation mechanism on Pt active sites is proposed, but various Pt active sites are observed in in-situ FTIR spectra of CO adsorption which can affect the formation of coke precursors. For instance, the linear adsorption peaks at 2,060 and 2,040 cm-1 correspond to high-coordination and low-coordination Pt species, respectively, and the bridge adsorption peak at 1,800 cm-1 [Supplementary Figure 11]. To investigate the influence of different Pt active phase structures on coke deposition, we designed multiple pulse experiments to observe the correlation between coke precursor generation and the structure of Pt active sites. After pulsing propane at 600 °C, the Pt/Sn-Al2O3 catalyst was cooled to 30 °C for CO adsorption analysis. Following complete CO desorption, the temperature was raised back to 600 °C for a new round of propane pulsing. The IR spectra after 1-10 rounds of propane pulsing [Figure 2] and in-situ FTIR spectra of CO adsorption after rounds 1-2 and 9-10 [Figure 3] were recorded.
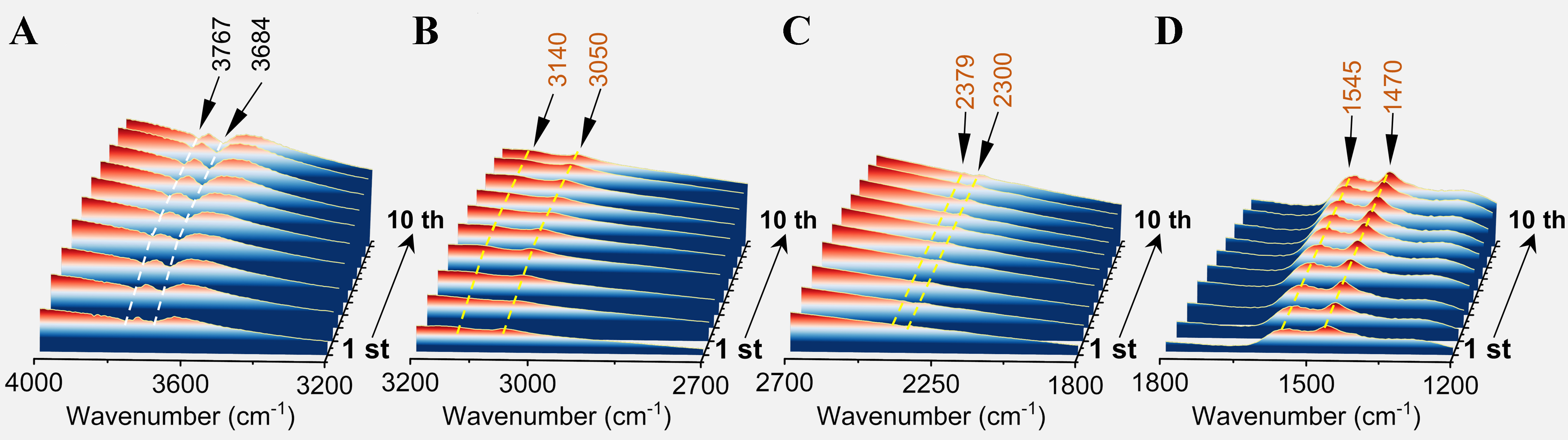
Figure 2. FTIR differential spectra of Pt/Sn-Al2O3 at 600 °C with different numbers of pulses of propane. (A) 4,000-3,200 cm-1; (B) 3,200-2,700 cm-1; (C) 2,700-1,800 cm-1; (D) 1,800-1,200 cm-1. The pulse experiments were carried out at 600 °C by pulses of pure propane (pulse volume = 0.50 cm3 STP) to the in situ cell, which was maintained under Ar flow (30 mL/min) between two successive pulses. The yellow dashed line represents the adsorption peak, while the white dashed line represents the inverted peak caused by the occupation of the adsorption site. FTIR: Fourier transform infrared spectroscopy; STP: standard temperature and pressure.
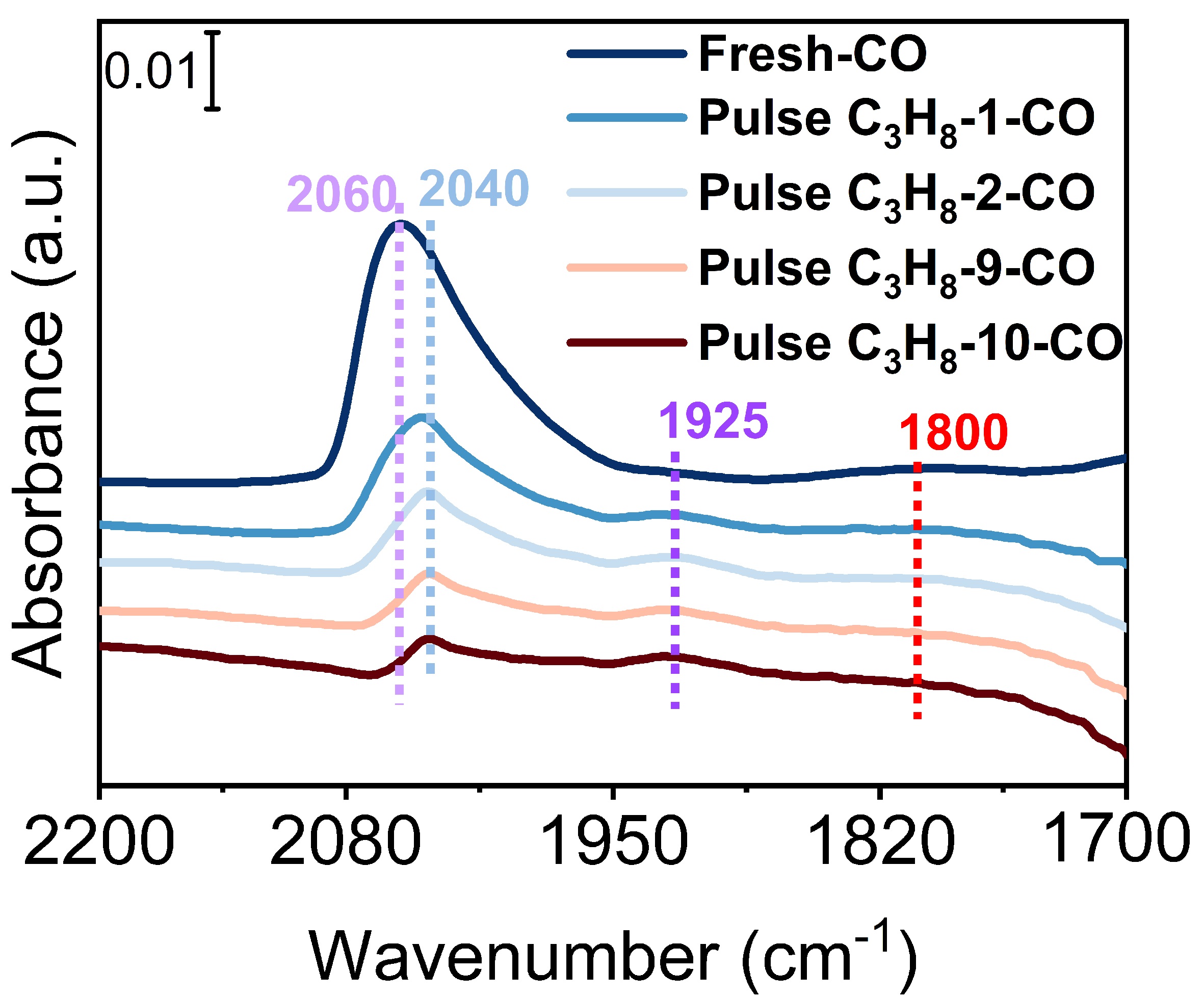
Figure 3. FTIR differential spectra of CO adsorption on the Pt/Sn-Al2O3 sample after pulse propane and reaction completion. After each pulse experiment, the sample was cooled to 30 °C, and the spectrum was collected after adsorption and desorption of CO. The temperature was raised to 600 °C for the next propane pulse experiment. The CO adsorption FTIR spectra of periods 1-2 and 9-10 were collected. FTIR: Fourier transform infrared spectroscopy.
Figure 2D shows that with increasing pulse numbers, the intensity of the absorption peaks at 1,470 and 1,545 cm-1 gradually rises, indicating the formation of monocyclic and polyaromatic hydrocarbon species. At the same time, the stretching vibrations of aromatic substituents or unsaturated hydrocarbons at 2,300-2,380 cm-1 (C=C=C) and the stretching vibration of unsaturated hydrocarbons at 2,190 cm-1 (C≡C) slightly increase [Figure 2C], attributed to aromatic substituents and unsaturated hydrocarbon species formed by deep dehydrogenation. This indicates that with the increasing number of pulses, monocyclic and polycyclic aromatic hydrocarbon precursors with alkyl substituents continuously form and deposit on the Pt sites. As the formation of coke precursor species, the gradual decrease in intensity within the 3,767-3,784 cm-1 hydroxyl region suggests that the surface hydroxyl groups of supports are covered by coke precursors [Figure 2A]. Combining the above analysis, a positive correlation is observed between the number of propane pulsing cycles and the content of aromatic species. The reduction in hydroxyl content suggests that coke deposition occurs not only at Pt sites but also on the surface of the Al2O3 support.
The intriguing observation from Figure 2D is that the rate of coke deposition during the first 1-2 pulsing cycles is significantly higher than during the 9-10 cycles. This demonstrates a shift in the coke deposition sites. Therefore, we attempt to elucidate the relationship between coke precursors and various Pt sites using in-situ FTIR spectra of CO adsorption [Figure 3]; the intensity of CO adsorption peaks on Pt active sites decreases as the number of pulsing cycles grows. This suggests that coke precursors poison parts of Pt sites. The linear CO adsorption peak for fresh catalysts is predominantly at 2,060 cm-1, shifting mainly to
This can also be confirmed by the correlation between PDH performance and coke deposition behavior of different Pt clusters [Figure 4]. The initial conversion of propane over the Pt/Al2O3 catalyst is 74.5%, which rapidly decreases to 49.2% after 1 h of reaction, a decline of 25.3% [Figure 4A]. This rapid decrease in activity is due to the extensive coke formed on surface Pt sites with saturated coordination [Figure 3, Supplementary Figure 11]. As coke extensively forms and covers these surface Pt sites with saturated coordination, the decreased rate of propane conversion slows down significantly after 1 h, indicating that the left Pt active sites have a strong capability to inhibit coke formation. These Pt active sites exist as low-coordination Pt sites with ultra-small size [Figure 3, Supplementary Figure 11]. As for the Pt/Sn-Al2O3 catalyst, the Sn can regulate Pt species to form more low-coordination Pt sites with ultra-small size, resulting in a slower deactivation of the Pt/Sn-Al2O3 catalyst. The initial conversion decreases from 33.2% to 22.5%, a reduction of only 10.7% [Figure 4B]. However, due to the electronic regulation of Pt by Sn, the conversion of Pt/Sn-Al2O3 catalyst is also reduced, which is significantly lower than the thermodynamic equilibrium conversion (~52%) under the same reaction condition [Supplementary Figure 7]. To further investigate the stability of the catalyst, a 100 h of PDH reaction was conducted for the Pt/Sn-Al2O3 catalyst
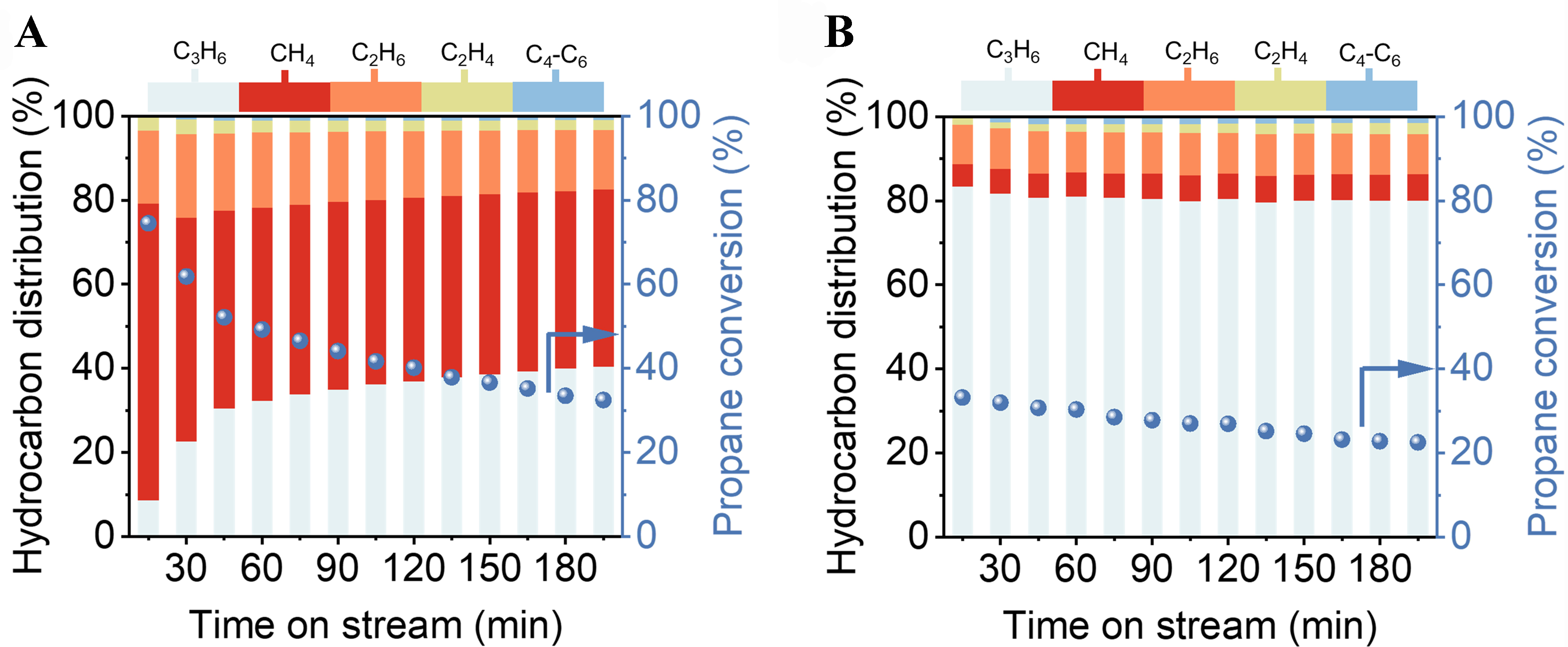
Figure 4. Product distribution of different catalysts as a function of time on stream during PDH for (A) Pt/Al2O3 and (B) Pt/Sn-Al2O3 samples. Dehydrogenation conditions: mcatalyst = 100 mg, C3H8/H2 = 1/1 (2.3 mL/min of propane and 2.3 mL/min of H2), Treduction =
Al2O3 carriers
Figure 5 shows the TG and corresponding differential TG (DTG) curves of catalysts after 3 h of PDH reaction. The degrees of weight loss in various catalysts increase in the order of Al2O3 (1.4%) < Sn-Al2O3 (1.9%) < Pt/Sn-Al2O3 (5.4%) < Pt/Al2O3 (18.5%) [Figure 5A]. The first weight loss peak within the 300-450 °C is attributed to coke at metal sites, while the peak within the 450-600 °C corresponds to coke on Al2O3 supports [Figure 5B]. Despite the lower coke content on the Al2O3, a peak in the high-temperature region suggests the formation of coke on the Al2O3 support. For the Sn-Al2O3 catalyst, weight loss is observed in both high- and low-temperature regions, with the high-temperature peak mainly due to coke on the support surface, and the low-temperature peak associated with monocyclic aromatic coke on SnOx species, as indicated by above FTIR analysis [Figure 1B]. The peaks for the Pt/Al2O3 catalyst are predominantly in the low-temperature region, suggesting that extensive coke deposition occurs on the Pt metal sites. The Pt/Sn-Al2O3 sample shows significant weight loss not only in the 300-400 °C range but also between 500-600 °C, confirming substantial coke accumulation on the Pt/Sn-Al2O3 support. Notably, the coke content on the pure Al2O3 sample is low, whereas the significantly severe coke deposition formed on the Al2O3 support in the Pt/Sn-Al2O3 under the same conditions, and there are distinct differences in the weight loss temperatures. These results further confirm that the introduction of Pt enhances coke deposition on the alumina support due to the synergistic effect of the dehydrogenation function of Pt and the acidity of the alumina support.
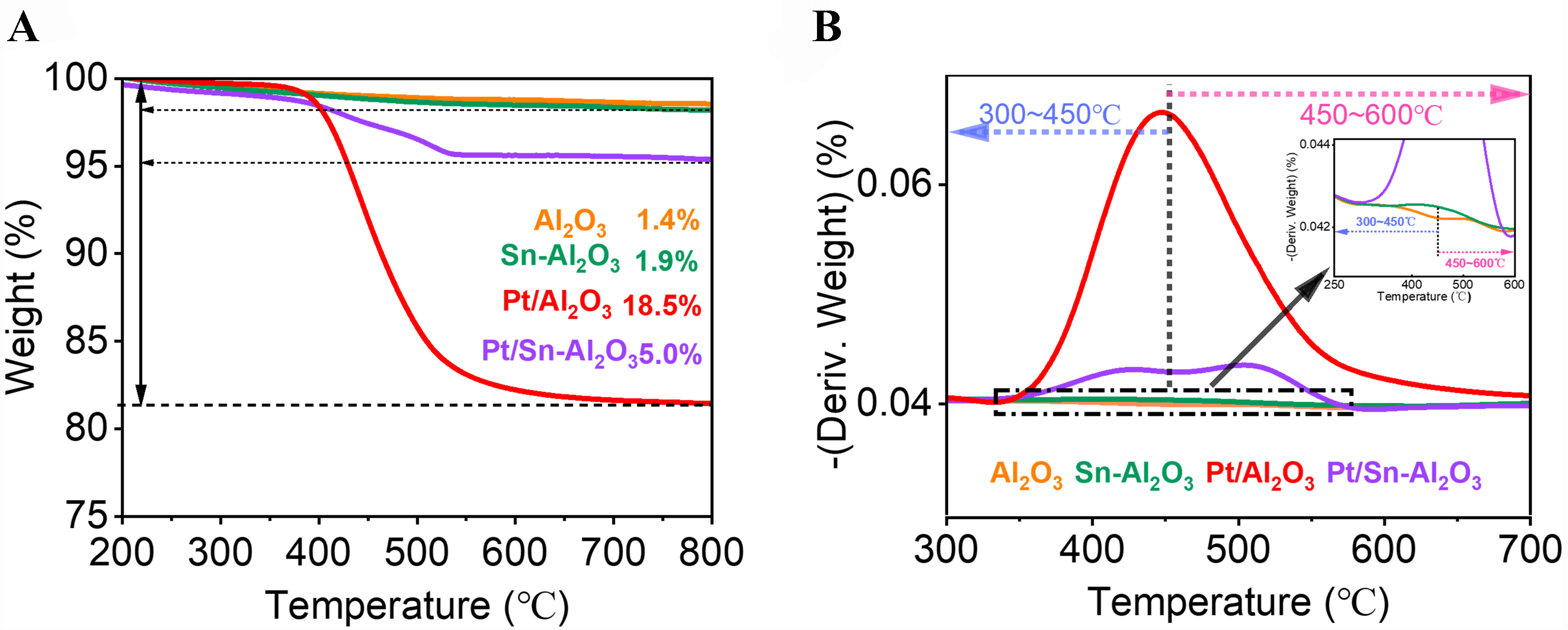
Figure 5. (A) TG profiles under air atmosphere and (B) corresponding DTG profiles of spent Al2O3, Sn-Al2O3, Pt/Al2O3 and Pt/Sn-Al2O3. The inset is the enlarged profiles at 250-600 °C. TG: Thermogravimetric; DTG: differential thermogravimetric.
The Raman spectroscopy was performed to further investigate the coke properties on various sites. Generally, the graphitic degree of coke can be reflected by the ID1/IG ratio (height ratio of D1 and G bands), with a smaller value indicating a higher graphitic degree [Supplementary Figure 14][67]. The ID1/IG ratios for Al2O3, Sn-Al2O3, Pt/Al2O3, Pt/Sn-Al2O3 after 3.3 h of reaction, and Pt/Sn-Al2O3 after 100 h of reaction are 0.96, 0.98, 0.71, 0.89, and 0.72, respectively. The ID1/IG ratios for Al2O3 and Sn-Al2O3 are significantly higher than those for the Pt-based catalysts. In addition, the H/C (mol ratio) values from elemental analysis in Supplementary Table 4 show that Al2O3 (15.85) and Sn-Al2O3 (19.59) have higher H/C ratios than the Pt-based catalysts (< 3.96), indicating higher hydrogen content in coke species. This suggests less graphitic coke on Al2O3 and Sn-Al2O3 than Pt/Al2O3 and Pt/Sn-Al2O3.
Confocal fluorescence microscopy was employed to detect the non-graphitized coke on the catalyst after reactions [Supplementary Figure 15]. The significantly higher fluorescence intensity for Al2O3 and Sn-Al2O3 than Pt-based catalysts indicates their lower graphitic degree, which agrees with the Raman result. As shown in the confocal fluorescence spectrum [Supplementary Figure 15F], the peaks located at 421-481, 486-556, 561-646 and 651-736 nm are attributed to the benzene, naphthalene, anthracene and phenanthrene, benzophenanthrene and triphenylene, and coronene. The types of coke on Al2O3 and Sn-Al2O3 are similar. Interestingly, the Sn-Al2O3 sample has a higher content of benzene species than other samples, indicating that SnOx sites are more prone to forming benzene species, which is consistent with the above FTIR results. The Pt/Al2O3 and Pt/Sn-Al2O3-100h samples did not show polycyclic coke species under fluorescence excitation, indicating that their coke species are mainly graphitized coke. These results suggest that the coke on Al2O3 and Sn-Al2O3 catalysts has a lower graphitic degree, compared with Pt-based catalysts.
To further explore the relationship between the Pt active phase, support acidity, and coke deposition on the support surface, Figure 6 presents the IR difference spectra after extensive isotope exchange reactions with H/D followed by pulsing with 500 Pa of propane for Al2O3 samples. As the reaction time increases, the intensity of the absorption peaks at 1,457-1,608 cm-1 gradually increases, indicating the formation of coke precursor species. Concurrently, the 2,400-2,800 cm-1 region, attributed to O–D stretching vibrations, shows a decreasing trend, while the 3,200-4,000 cm-1 region, corresponding to O–H stretching vibrations, tends to increase. This suggests that the O–D groups on the alumina support participate in the dehydrogenation of reactant molecules, leading to the formation of coke. Consequently, D atoms on the support surface are replaced by H, forming O–H. These data indicate that deep dehydrogenation and cyclization to form aromatic coke precursors occur on the Al2O3 surface, further confirming that the support contributes to the formation of coke precursors. In addition, the deactivation constant for the catalysts shows that the deactivation constant of Al2O3 is slightly higher than that of the catalysts containing Pt [Supplementary Table 5]. This indicates that the active sites on Al2O3 can accumulate coke, leading to catalyst deactivation.
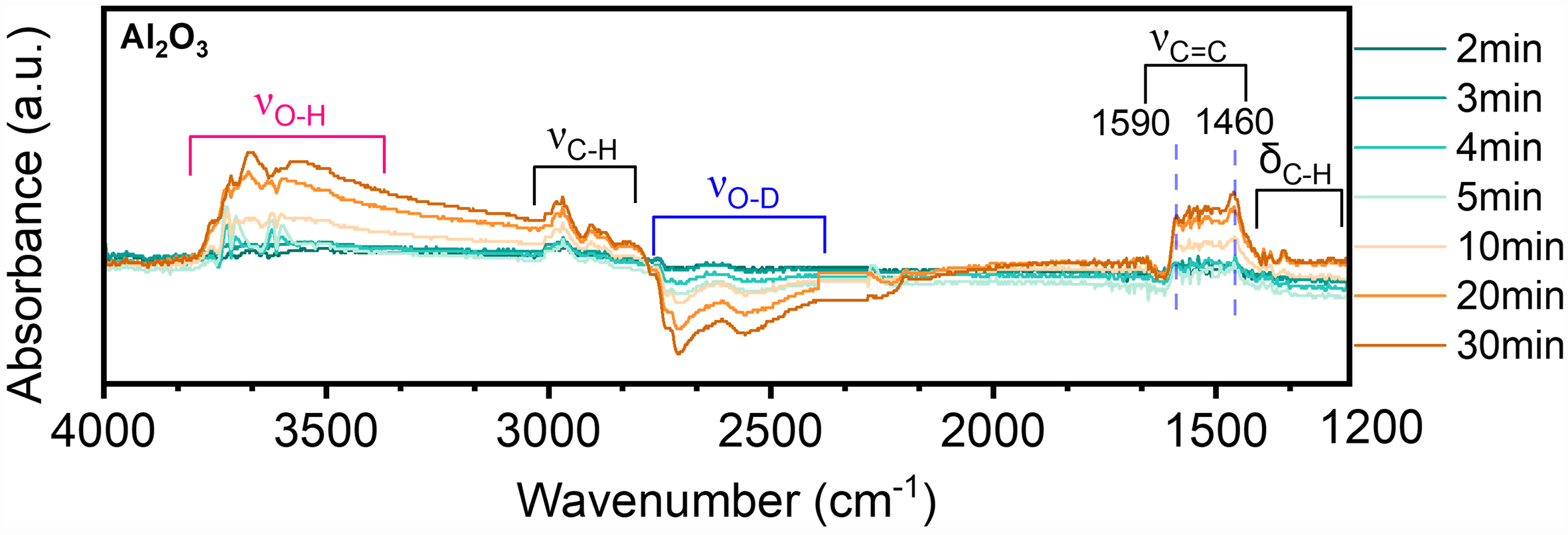
Figure 6. FTIR differential spectra following propane pulse adsorption for Al2O3 after D2O replacement. The pulse experiments were carried out at 600 °C by pulses of pure propane (pulse volume = 0.50 cm3 STP) to the in situ cell at Ar (30 mL/min) atmosphere. FTIR: Fourier transform infrared spectroscopy; STP: standard temperature and pressure.
The effect of hydrogen co-feed
It is well known that the introduction of H2 can suppress the coke deposits in PDH. Therefore, we pulsed pure propane or C3H8/H2 mixture (volume ratio = 1:1) (pulse volume = 0.50 cm3 STP) to the in situ cell under Ar flow (30 mL/min), and continuously recorded FTIR spectra for 5 min. Figure 7 presents a comparative FTIR difference spectrum of propane and a propane-hydrogen gas mixture, further exploring the impact of H2 on the formation of coke precursor species on the Pt/Sn-Al2O3 catalyst. The absorption peaks attributed to aromatic hydrocarbons ν (C=C) in 1,470-1,600 cm-1 show a significant decrease in intensity [Figure 7B and D], indicating that the introduction of H2 effectively inhibits the formation of aromatic and unsaturated hydrocarbons. Additionally, an observation of the νm (C–H) vibrational peak in the aromatic region of 3,050-3,140 cm-1 further suggests that the aromatic species are dominated by a higher H/C ratio [Figure 7A and C]. The introduction of H2 reduces the content and changes the types of coke precursor species.
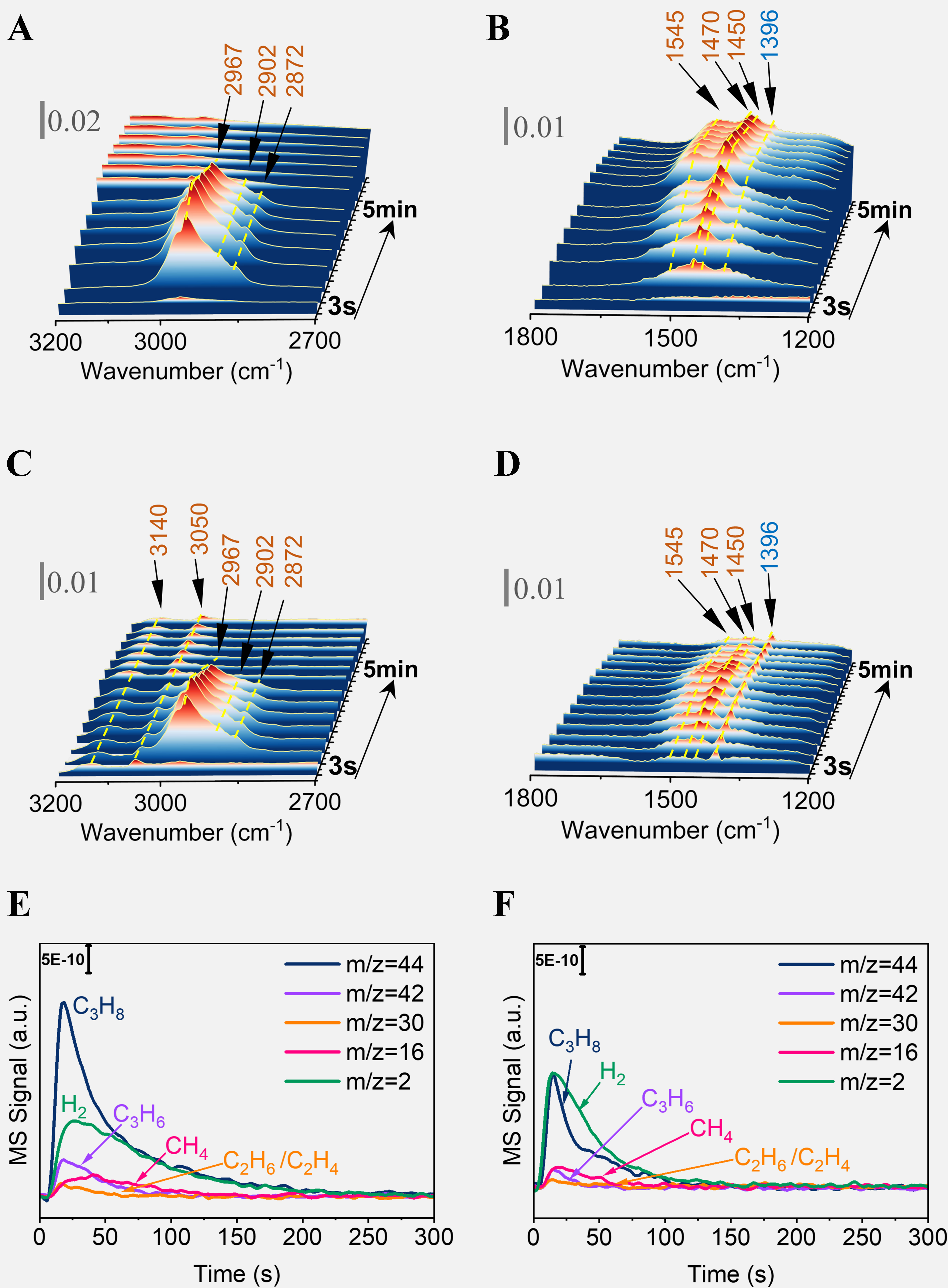
Figure 7. FTIR differential spectra following (A and B) pure C3H8 and (C and D) C3H8/H2 mixture (volume ratio = 1:1) pulse adsorption for Pt/Sn-Al2O3, and corresponding mass spectrum of (E) pure C3H8 and (F) C3H8/H2 mixture pulse. The pulse experiments were carried out at 600 °C by pulses of pure propane and C3H8/H2 mixture (pulse volume = 0.50 cm3 STP) to the in situ cell at Ar
In Supplementary Figure 16, in-situ FTIR spectra of CO adsorption were utilized to investigate the effect of feed composition on Pt sites. The area of the CO adsorption peak at Pt sites in the mixed-feed sample exhibits a lesser decline compared to the propane-only feed, indicating that the presence of H2 inhibits the poisoning of Pt sites by coke deposition. In the mass spectrometry (MS) analysis presented in Figure 7C and D, a significant amount of propylene desorption is detected in the MS when C3H8 is fed alone, coupled with an increase in the intensity of the coke deposition IR peaks, suggesting that deep dehydrogenation on the catalyst leads to coke formation and poisons the Pt sites. Conversely, when the mixed feed is analyzed, a higher content of hydrogenolysis product CH4 is detected, along with a decrease in the intensity of the coke IR peaks, indicating that the coke precursor species on the Pt sites undergo hydrogenolysis, thereby inhibiting the poisoning of Pt sites by coke. The introduction of hydrogen effectively controls and adjusts the content and types of coke precursor species while also preventing the poisoning of Pt sites by coke.
CONCLUSIONS
The formation mechanisms of coke at Al2O3, Sn, Pt and Pt-Sn sites of Al2O3 supported PtSn catalysts in PDH are clarified. Compared to Pt and Pt-Sn sites, the coke initiation capability of Al2O3 and Sn is significantly weaker. The tri-coordinated aluminum with Lewis acidity in Al2O3 is the primary site for coke formation, where propane undergoes excessive dehydrogenation to form propylidene and propylidyne, which further cyclize to form monocyclic and polycyclic aromatic coke precursors. The coke precursors at SnOx sites differ from those on Al2O3, where the propyl species only forms monocyclic aromatic coke precursors after excessive dehydrogenation. For the Pt/Sn-Al2O3 catalyst, the strong dehydrogenation function and the interaction between Pt and supports trigger the complex coke formation mechanism. The coke formation is mainly ascribed to the following three factors: the structures of Pt-Sn clusters, active hydrogen species, Al2O3 support under the influence of active hydrogen species. The surface Pt sites with saturated coordination are prone to coke deposition, while the low-coordination Pt sites with ultra-small size are found to be highly resistant to coke formation in the PDH reaction. The strong dehydrogenation ability of Pt and the active hydrogen species play a vital role in the formation of coke precursors on Al2O3. Hydrogen co-feed also affects the formation of coke precursor. Hydrogen co-feed significantly inhibits the formation of coke precursors and leads to the formation of aromatic hydrocarbon coke precursors with higher H/C ratios. This work can provide the theoretical basis for the development of PDH catalysts with excellent coking resistance performance, and the established research method of coke is expected to be applied to more catalytic research fields.
DECLARATIONS
Authors’ contributions
Designed, prepared and revised the manuscript: Jiao, J., Yang, Y., Song, L.
Discussion and preparation of the manuscript: Jiao, J., Yang, Y., Yuan, M., Tang, X., Shi, M., He, K., Zhang, H., Bi, Y., Qin, Y., Song, L.
Availability of data and materials
The detailed materials and methods in the experiment were listed in the Supplementary Materials. Other raw data that support the findings of this study are available from the corresponding author upon reasonable request.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (No. U1908203; No. U20A20120) and Basic Scientific Research Project of Liaoning Province Department of Education (No. JYTQN2023342).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
1. Song, S.; Yang, K.; Zhang, P.; et al. Silicalite-1 stabilizes Zn-hydride species for efficient propane dehydrogenation. ACS. Catal. 2022, 12, 5997-6006.
2. Hou, W.; Lin, K.; Zhang, X.; et al. Highly stable and selective Pt/TS-1 catalysts for the efficient nonoxidative dehydrogenation of propane. Chem. Eng. J. 2023, 474, 145648.
3. Festa, G.; Contaldo, P.; Martino, M.; Meloni, E.; Palma, V. Modeling the selectivity of hydrotalcite-based catalyst in the propane dehydrogenation reaction. Ind. Eng. Chem. Res. 2023, 62, 16622-37.
4. Phadke, N. M.; Mansoor, E.; Bondil, M.; Head-Gordon, M.; Bell, A. T. Mechanism and kinetics of propane dehydrogenation and cracking over Ga/H-MFI prepared via vapor-phase exchange of H-MFI with GaCl3. J. Am. Chem. Soc. 2019, 141, 1614-27.
5. Nakaya, Y.; Hirayama, J.; Yamazoe, S.; Shimizu, K. I.; Furukawa, S. Single-atom Pt in intermetallics as an ultrastable and selective catalyst for propane dehydrogenation. Nat. Commun. 2020, 11, 2838.
6. Ma, Y.; Chen, X.; Guan, Y.; et al. Skeleton-Sn anchoring isolated Pt site to confine subnanometric clusters within *BEA topology. J. Catal. 2021, 397, 44-57.
7. Shi, L.; Deng, G. M.; Li, W. C.; et al. Al2O3 nanosheets rich in pentacoordinate Al3+ ions stabilize Pt-Sn clusters for propane dehydrogenation. Angew. Chem. Int. Ed. Engl. 2015, 54, 13994-8.
8. Ma, Y.; Song, S.; Liu, C.; et al. Germanium-enriched double-four-membered-ring units inducing zeolite-confined subnanometric Pt clusters for efficient propane dehydrogenation. Nat. Catal. 2023, 6, 506-18.
9. Wei, S.; Dai, H.; Long, J.; et al. Nonoxidative propane dehydrogenation by isolated Co2+ in BEA zeolite: dealumination-determined key steps of propane C–H activation and propylene desorption. Chem. Eng. J. 2023, 455, 140726.
10. Wan, H.; Qian, L.; Gong, N.; et al. Size-dependent structures and catalytic properties of supported bimetallic PtSn catalysts for propane dehydrogenation reaction. ACS. Catal. 2023, 13, 7383-94.
11. Wang, P.; Senftle, T. P. Modeling phase formation on catalyst surfaces: coke formation and suppression in hydrocarbon environments. AIChE. J. 2021, 67, e17454.
12. Zhang, Y.; Aly, M. Effect of CO2 on activity and coke formation over gallium-based catalysts for propane dehydrogenation. Appl. Catal. A. Gen. 2022, 643, 118795.
13. Sun, M.; Hu, Z.; Wang, H.; Suo, Y.; Yuan, Z. Design strategies of stable catalysts for propane dehydrogenation to propylene. ACS. Catal. 2023, 13, 4719-41.
14. Chen, S.; Chang, X.; Sun, G.; et al. Propane dehydrogenation: catalyst development, new chemistry, and emerging technologies. Chem. Soc. Rev. 2021, 50, 3315-54.
15. Xiao, L.; Shan, Y.; Sui, Z.; et al. Beyond the reverse Horiuti–Polanyi mechanism in propane dehydrogenation over Pt catalysts. ACS. Catal. 2020, 10, 14887-902.
16. Xiong, H.; Lin, S.; Goetze, J.; et al. Thermally stable and regenerable platinum–tin clusters for propane dehydrogenation prepared by atom trapping on ceria. Angew. Chem. 2017, 129, 9114-9.
17. Zhang, W.; Wang, H.; Jiang, J.; et al. Size dependence of Pt catalysts for propane dehydrogenation: from atomically dispersed to nanoparticles. ACS. Catal. 2020, 10, 12932-42.
18. Gao, X.; Yao, Z.; Li, W.; et al. Calcium-modified PtSn/Al2O3 catalyst for propane dehydrogenation with high activity and stability. ChemCatChem 2023, 15, e202201691.
19. Yu, Q.; Yu, T.; Chen, H.; Fang, G.; Pan, X.; Bao, X. The effect of Al3+ coordination structure on the propane dehydrogenation activity of Pt/Ga/Al2O3 catalysts. J. Energy. Chem. 2020, 41, 93-9.
20. Kwak, J. H.; Hu, J.; Mei, D.; et al. Coordinatively unsaturated Al3+ centers as binding sites for active catalyst phases of platinum on gamma-Al2O3. Science 2009, 325, 1670-3.
21. Li, X.; Li, J.; Yuan, M.; et al. Effect of chlorine on the performance of Cr-K/γ-Al2O3 catalyst for n-hexane dehydrogenation. Appl. Catal. A. Gen. 2023, 664, 119322.
22. Sun, G.; Zhao, Z. J.; Li, L.; et al. Metastable gallium hydride mediates propane dehydrogenation on H2 co-feeding. Nat. Chem. 2024, 16, 575-83.
23. Wang, Z.; Chen, Y.; Mao, S.; et al. Chemical insight into the structure and formation of coke on PtSn alloy during propane dehydrogenation. Adv. Sustain. Syst. 2020, 4, 2000092.
24. Zhao, Z. J.; Wu, T.; Xiong, C.; et al. Hydroxyl-mediated non-oxidative propane dehydrogenation over VOx/γ-Al2O3 catalysts with improved stability. Angew. Chem. Int. Ed. Engl. 2018, 57, 6791-5.
25. Sun, G.; Zhao, Z. J.; Mu, R.; et al. Breaking the scaling relationship via thermally stable Pt/Cu single atom alloys for catalytic dehydrogenation. Nat. Commun. 2018, 9, 4454.
26. Biloen, P. Catalytic dehydrogenation of propane to propene over platinum and platinum-gold alloys. J. Catal. 1977, 50, 77-86.
27. Sattler, J. J.; Ruiz-Martinez, J.; Santillan-Jimenez, E.; Weckhuysen, B. M. Catalytic dehydrogenation of light alkanes on metals and metal oxides. Chem. Rev. 2014, 114, 10613-53.
28. Yuan, Y.; Brady, C.; Lobo, R. F.; Xu, B. Understanding the correlation between Ga speciation and propane dehydrogenation activity on Ga/H-ZSM-5 catalysts. ACS. Catal. 2021, 11, 10647-59.
29. Phadke, N. M.; Mansoor, E.; Head-gordon, M.; Bell, A. T. Mechanism and kinetics of light alkane dehydrogenation and cracking over isolated Ga species in Ga/H-MFI. ACS. Catal. 2021, 11, 2062-75.
30. Li, M.; Yin, W.; Pan, J.; et al. Hydrogen spillover as a promising strategy for boosting heterogeneous catalysis and hydrogen storage. Chem. Eng. J. 2023, 471, 144691.
31. Niu, X.; Zhao, R.; Han, Y.; Zhang, X.; Wang, Q. Highly dispersed platinum clusters anchored on hollow ZSM-5 zeolite for deep hydrogenation of polycyclic aromatic hydrocarbons. Fuel 2022, 326, 125021.
32. Wang, G.; Li, C.; Shan, H. Catalytic dehydrogenation of isobutane over a Ga2O3/ZnO interface: reaction routes and mechanism. Catal. Sci. Technol. 2016, 6, 3128-36.
33. Dewangan, N.; Ashok, J.; Sethia, M.; et al. Cobalt-based catalyst supported on different morphologies of alumina for non-oxidative propane dehydrogenation: effect of metal support interaction and lewis acidic sites. ChemCatChem 2019, 11, 4923-34.
34. Soma, Y. Infrared spectra of ethylene adsorbed on transition metals at low temperatures and hydrogenation of the adsorbed species. J. Catal. 1979, 59, 239-47.
35. Ko, M. K.; Frei, H. Millisecond FT-IR spectroscopy of surface intermediates of C2H4 hydrogenation over Pt/Al2O3 catalyst under reaction conditions. J. Phys. Chem. B. 2004, 108, 1805-8.
36. Arena, F.; Dario, R.; Parmaliana, A. A characterization study of the surface acidity of solid catalysts by temperature programmed methods. Appl. Catal. A. 1998, 170, 127-37.
37. Buzzoni, R.; Bordiga, S.; Ricchiardi, G.; Lamberti, C.; Zecchina, A.; Bellussi, G. Interaction of pyridine with acidic (H-ZSM5, H-β, H-MORD zeolites) and superacidic (H-nafion membrane) systems: an IR investigation. Langmuir 1996, 12, 930-40.
38. Cholewinski, M. C.; Dixit, M.; Mpourmpakis, G. Computational study of methane activation on γ-Al2O3. ACS. Omega. 2018, 3, 18242-50.
39. Dixit, M.; Kostetskyy, P.; Mpourmpakis, G. Structure–activity relationships in alkane dehydrogenation on γ-Al2O3: site-dependent reactions. ACS. Catal. 2018, 8, 11570-8.
40. Otroshchenko, T.; Sokolov, S.; Stoyanova, M.; et al. ZrO2-based alternatives to conventional propane dehydrogenation catalysts: active sites, design, and performance. Angew. Chem. Int. Ed. Engl. 2015, 54, 15880-3.
41. Li, C.; Guo, X.; Shang, Q.; et al. Defective TiO2 for propane dehydrogenation. Ind. Eng. Chem. Res. 2020, 59, 4377-87.
42. Wang, P.; Xu, Z.; Wang, T.; Yue, Y.; Bao, X.; Zhu, H. Unmodified bulk alumina as an efficient catalyst for propane dehydrogenation. Catal. Sci. Technol. 2020, 10, 3537-41.
43. Leclerc, H.; Vimont, A.; Lavalley, J. C.; et al. Infrared study of the influence of reducible iron(III) metal sites on the adsorption of CO, CO2, propane, propene and propyne in the mesoporous metal-organic framework MIL-100. Phys. Chem. Chem. Phys. 2011, 13, 11748-56.
44. Čapek, L.; Novoveská, K.; Sobalík, Z.; Wichterlová, B.; Cider, L.; Jobson, E. Cu-ZSM-5 zeolite highly active in reduction of NO with decane under water vapor presence. Appl. Catal. B. Environ. 2005, 60, 201-10.
45. Hadjiivanov, K. Chapter two - Identification and characterization of surface hydroxyl groups by infrared spectroscopy. Adv. Catal. 2014, 57, 99-318.
46. Vazhnova, T.; Rigby, S. P.; Lukyanov, D. B. Benzene alkylation with ethane in ethylbenzene over a PtH-MFI catalyst: kinetic and IR investigation of the catalyst deactivation. J. Catal. 2013, 301, 125-33.
47. Madeira, F. F.; Vezin, H.; Gnep, N. S.; Magnoux, P.; Maury, S.; Cadran, N. Radical species detection and their nature evolution with catalyst deactivation in the ethanol-to-hydrocarbon reaction over HZSM-5 zeolite. ACS. Catal. 2011, 1, 417-24.
48. Song, C.; Liu, K.; Zhang, D.; et al. Effect of cofeeding n-butane with methanol on aromatization performance and coke formation over a Zn loaded ZSM-5/ZSM-11 zeolite. Appl. Catal. A. Gen. 2014, 470, 15-23.
49. Roy, S.; Bakhmutsky, K.; Mahmoud, E.; Lobo, R. F.; Gorte, R. J. Probing lewis acid sites in Sn-beta zeolite. ACS. Catal. 2013, 3, 573-80.
50. Zhu, Y.; An, Z.; Song, H.; Xiang, X.; Yan, W.; He, J. Lattice-confined Sn (IV/II) stabilizing raft-like Pt clusters: high selectivity and durability in propane dehydrogenation. ACS. Catal. 2017, 7, 6973-8.
51. Wang, F.; Xiao, W.; Gao, L.; Xiao, G. Enhanced performance of glycerol to aromatics over Sn-containing HZSM-5 zeolites. RSC. Adv. 2016, 6, 42984-93.
52. Karge, H.; Nießen, W.; Bludau, H. In-situ FTIR measurements of diffusion in coking zeolite catalysts. Appl. Catal. A. Gen. 1996, 146, 339-49.
53. Wong, K. S.; Vazhnova, T.; Rigby, S. P.; Lukyanov, D. B. Temperature effects in benzene alkylation with ethane into ethylbenzene over a PtH-MFI bifunctional catalyst. Appl. Catal. A. Gen. 2013, 454, 137-44.
54. van den Brand J, Blajiev O, Beentjes PC, Terryn H, de Wit JH. Interaction of ester functional groups with aluminum oxide surfaces studied using infrared reflection absorption spectroscopy. Langmuir 2004, 20, 6318-26.
55. Zaki, M. I.; Hasan, M. A.; Al-sagheer, F. A.; Pasupulety, L. Surface chemistry of acetone on metal oxides: IR observation of acetone adsorption and consequent surface reactions on silica−alumina versus silica and alumina. Langmuir 2000, 16, 430-6.
56. Airaksinen, S.; Banares, M.; Krause, A. In situ characterisation of carbon-containing species formed on chromia/alumina during propane dehydrogenation. J. Catal. 2005, 230, 507-13.
57. Finocchio, E.; Busca, G.; Lorenzelli, V.; Willey, R. The activation of hydrocarbon CH bonds over transition metal oxide catalysts: a FTIR study of hydrocarbon catalytic combustion over MgCr2O4. J. Catal. 1995, 151, 204-15.
58. Deng, X.; Qin, B.; Liu, R.; et al. Zeolite-encaged isolated platinum ions enable heterolytic dihydrogen activation and selective hydrogenations. J. Am. Chem. Soc. 2021, 143, 20898-906.
59. Pei, Y.; Qi, Z.; Goh, T. W.; et al. Intermetallic structures with atomic precision for selective hydrogenation of nitroarenes. J. Catal. 2017, 356, 307-14.
60. Vaarkamp, M.; Miller, J. T.; Modica, F. S.; Koningsberger, D. C. On the relation between particle morphology, structure of the metal-support interface, and catalytic properties of Pt/γ-Al2O3. J. Catal. 1996, 163, 294-305.
61. Bazin, P.; Saur, O.; Lavalley, J. C.; Daturi, M.; Blanchard, G. FT-IR study of CO adsorption on Pt/CeO2: characterisation and structural rearrangement of small Pt particles. Phys. Chem. Chem. Phys. 2005, 7, 187.
62. Liu, L.; Lopez-haro, M.; Lopes, C. W.; et al. Structural modulation and direct measurement of subnanometric bimetallic PtSn clusters confined in zeolites. Nat. Catal. 2020, 3, 628-38.
63. Han, L.; Meng, Q.; Wang, D.; et al. Interrogation of bimetallic particle oxidation in three dimensions at the nanoscale. Nat. Commun. 2016, 7, 13335.
64. Kim, T.; Kim, J.; Song, H. C.; et al. Catalytic synergy on PtNi bimetal catalysts driven by interfacial intermediate structures. ACS. Catal. 2020, 10, 10459-67.
65. Wang, C. M.; Schreiber, D. K.; Olszta, M. J.; Baer, D. R.; Bruemmer, S. M. Direct in Situ TEM observation of modification of oxidation by the injected vacancies for Ni-4Al alloy using a microfabricated nanopost. ACS. Appl. Mater. Interfaces. 2015, 7, 17272-7.
66. Allian, A. D.; Takanabe, K.; Fujdala, K. L.; et al. Chemisorption of CO and mechanism of CO oxidation on supported platinum nanoclusters. J. Am. Chem. Soc. 2011, 133, 4498-517.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Author Biographies
























Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].