

Electrosynthesis reactions with divided cells: unlocking potentials in organic synthesis


Abstract
Electrosynthesis reactions have become an important field of study due to the increasing demand for sustainable and environmentally friendly chemical processes. Using a divided cell in electrosynthesis has shown promising results in selectivity, efficiency, and scalability. In this review, we discuss the principles and advantages of using divided cells in electrosynthesis reactions, focusing on their application in producing organic compounds. We also consider several factors that influence the performance of divided cells, such as the choice of electrode materials, membrane type, and operating conditions. Furthermore, we evaluate the challenges and limitations associated with divided cell electrosynthesis, including the need for high current densities and the management of gas evolution.

Keywords
INTRODUCTION
Electrosynthetic reactions have gained significant attention in recent years due to their potential for sustainable and eco-friendly chemical production[1]. The shift from redox reagents to electricity, a renewable energy source, enables selective and efficient transformations of organic and inorganic compounds[2], reduces fossil fuel use, and minimizes waste[3,4], though the environmental impact of manufacturing materials such as electrodes and membranes demands further attention[5,6].
Electrosynthetic methods have proven to be innovative for obtaining high-value-added organic products[7]. Although many of these methodologies are widely developed in undivided cells, the use of divided cells has gained attention for offering advantageous alternatives in certain systems, particularly for controlling selectivity and mitigating issues such as electrode passivation and reduced reaction rates[8,9], in addition to being explored in various applications such as water treatment[10] and energy storage[11,12].
Assembly of divided cells for electrosynthesis
Prototypes of divided cells emerged more than a century ago as a way to prevent interference between cathodic and anodic reactions[13,14]. Divided cells employ a semipermeable membrane that allows conductivity through the selective passage of ions, but not the starting materials or products, enabling electrolysis without mixing the cathodic and anodic solutions [Figure 1]. The compartments can be divided using low-porosity ceramic frits or polymeric membranes, such as Nafion[15,16].
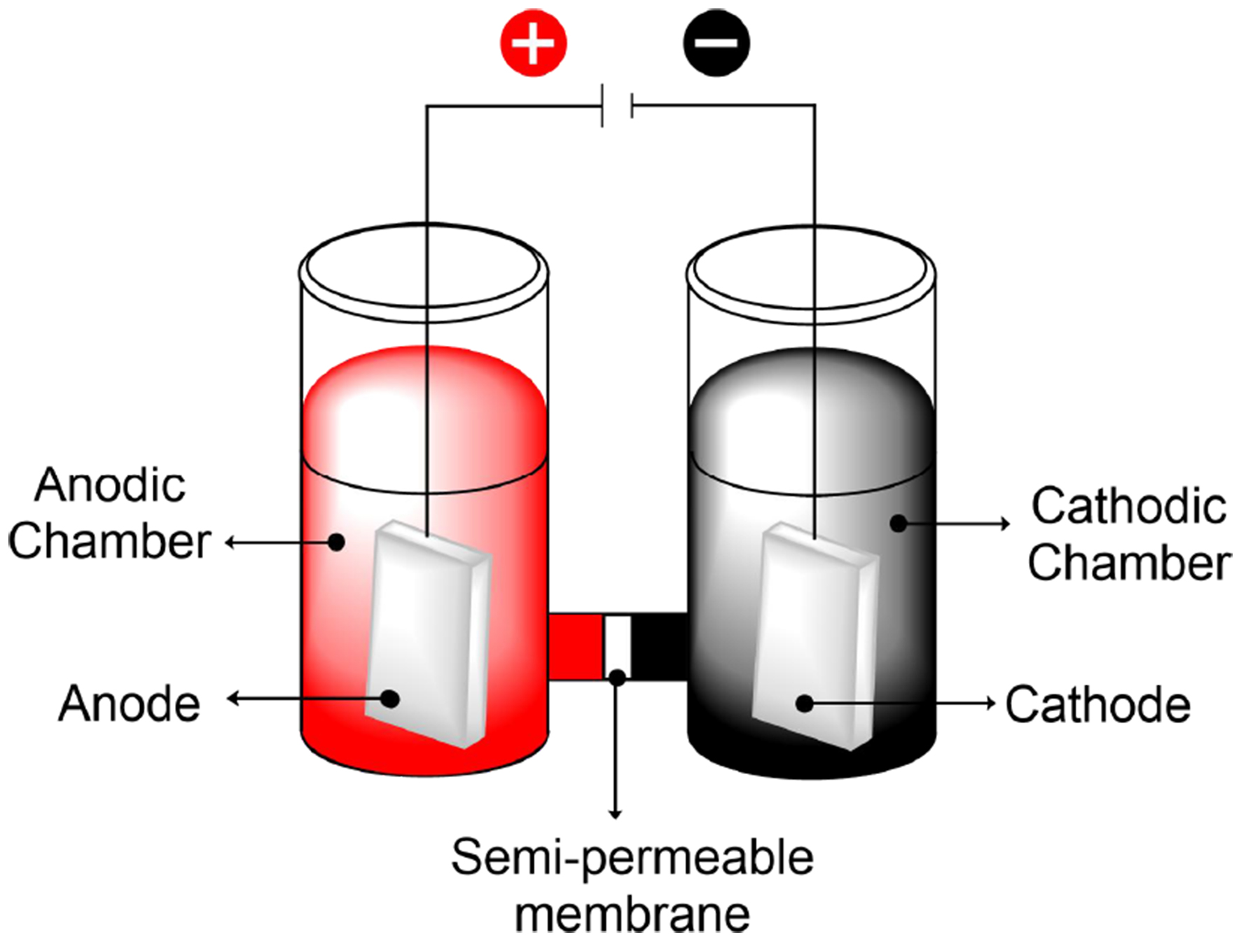
Figure 1. Schematically divided electrochemical cell.
Although divided cells have a more complex design compared to undivided cells, they can offer a viable alternative when the use of undivided cells becomes impractical. Divided cells provide notable advantages, such as increased selectivity, higher current efficiency, and the prevention of undesirable side reactions. Since the cathode and anode compartments are physically separated, side redox reactions are avoided. This allows independent optimization of the reactions at each electrode, with precise control of reaction conditions and efficient separation of products [Figure 2][16]. One of the key challenges in the industrial adoption of divided cell electrosynthesis lies in achieving high current densities, which are essential for economically viable production scales. Industrially relevant current densities typically range from 0.3 to
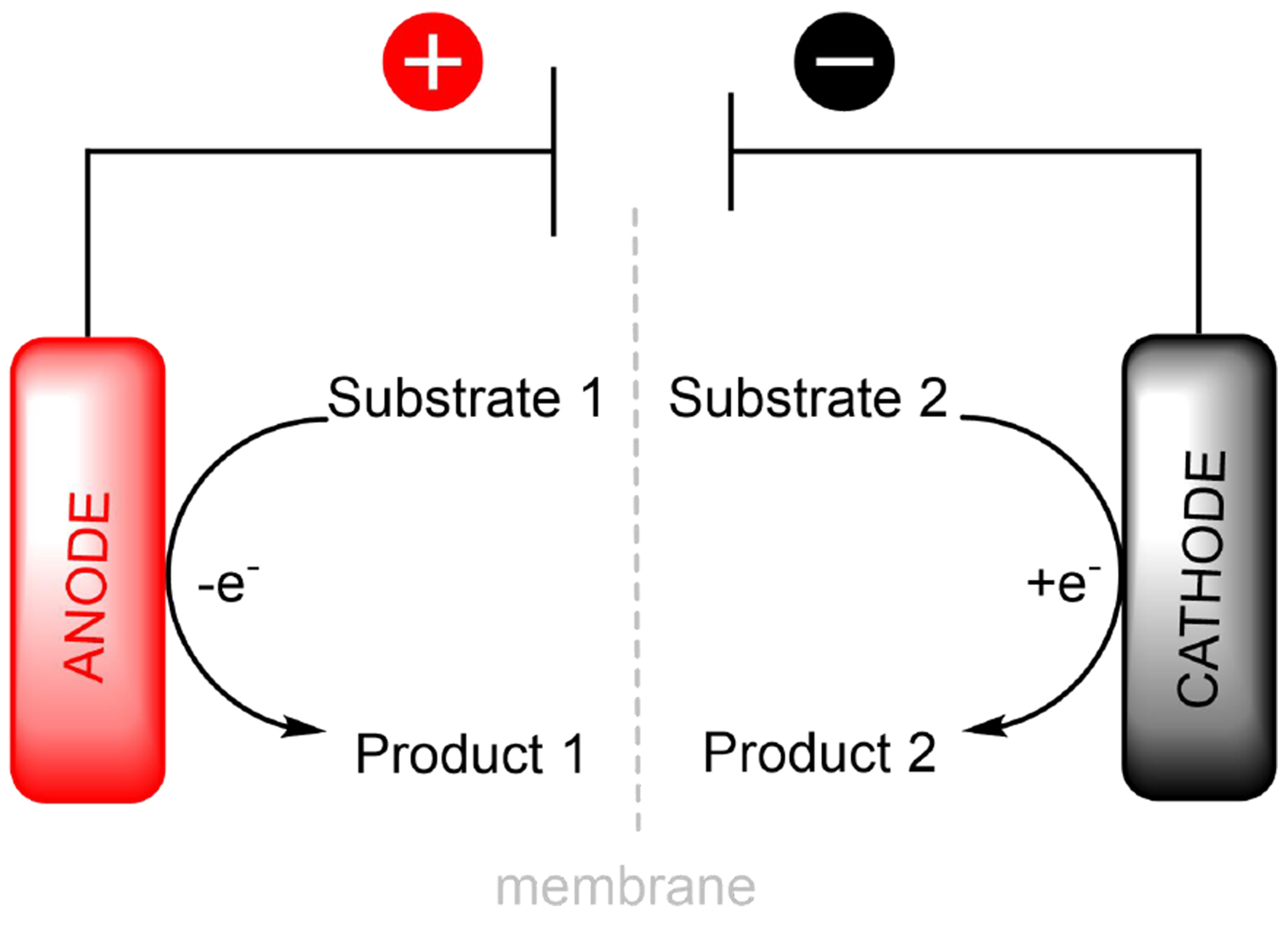
Figure 2. Electrolysis in a divided cell.
Paired electrode reactions represent one possible configuration involving simultaneous redox processes where oxidation and reduction take place in tandem, connected through an electron flow. The selection of compatible redox pairs is crucial to optimize electron transfer (ET) and minimize energy losses, thereby enhancing system efficiency. These reactions stand out for their ability to improve selectivity, reduce undesirable byproducts, and play a significant role in organic synthesis, particularly when renewable energy sources are employed, aligning with sustainable practices[18].
Overall, electrolysis can be described as comprising two half-cell reactions: anodic oxidation and cathodic reduction, which are interconnected through a paired process. As illustrated in Figure 2, due to the separation of the two compartments, there is no contact between substrate 1 and product 1 with the cathode. Additionally, the reaction that forms product 2 at the cathode can be productive, utilizing electrical current at both electrodes in a paired electrolysis, and there are no issues with separating the two products. There are some examples of paired electrolysis in divided cells described in the literature[19-22], but mostly exploring only one of the reactions. Formally, this is not so valuable as it reduces energy efficiency with only part of the energy being used for the formation of products[23]. This limitation highlights the need for further optimization strategies, such as reaction conditions, to enhance the energy utilization in paired electrolysis setups.
Configuration of divided cells
Electrosynthesis involves several variables and particularities along with the cell design, such as the electrode material, current, electric potential, solvent, and electrolytes. Electrochemical procedures are based on ET, either from the electrode to the substrate or from the substrate to the electrode, and there are different manners by which this transfer can occur. Direct electrolysis (non-mediated) occurs when the substrate is oxidized or reduced directly at the electrode, whether it is inert or activated. In contrast, indirect (mediated) electrolysis involves the use of an intermediate species, such as a mediator or redox catalyst, to transfer electrons between the electrode and the substrate [Figure 3].
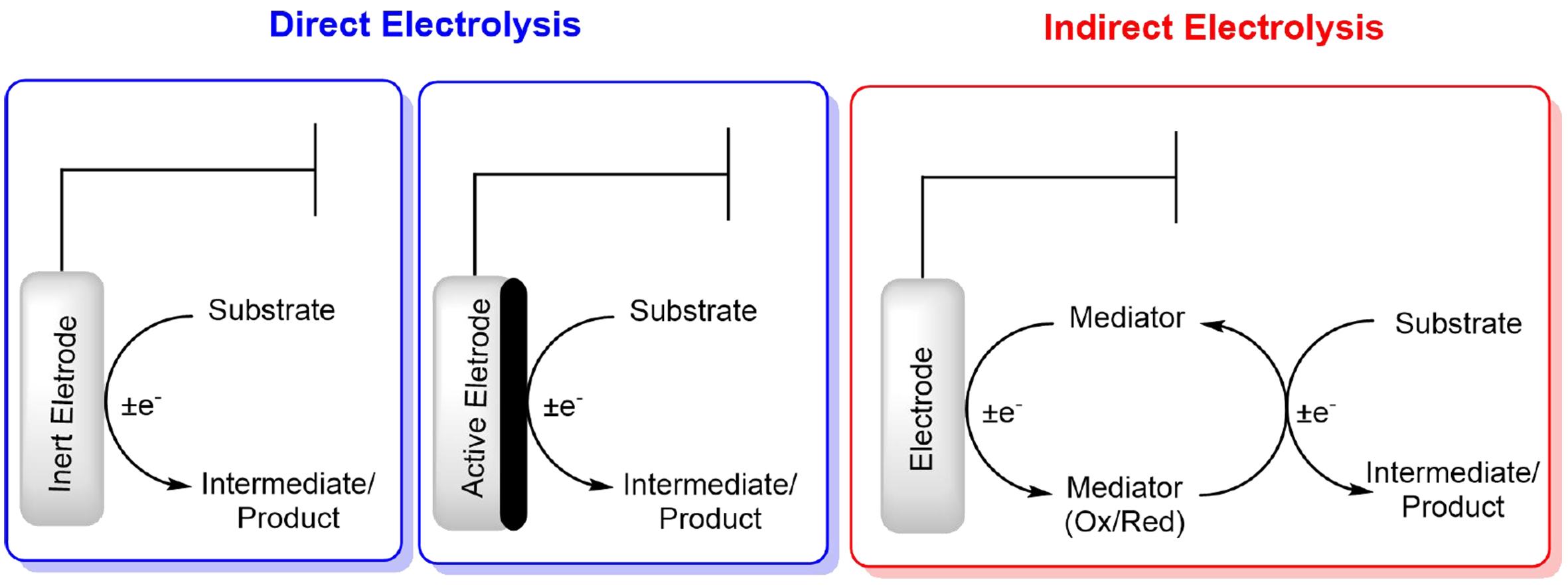
Figure 3. Direct and indirect ET in electrosynthesis. ET: Electron transfer.
In direct electrolysis processes, inert electrodes such as those composed of platinum, graphite, or boron-doped diamond are widely employed due to their chemical stability and ability to resist high current densities without promoting side reactions. These materials ensure efficient and direct ET (DET) between the electrode and the target substrate. Conversely, indirect electrolysis processes using active electrodes require electrodes specifically designed to facilitate the generation of reactive intermediate species that subsequently interact with the substrate. Electrodes based on metal oxides (e.g., manganese, nickel, and iridium oxides) or surfaces functionalized with polymer or redox catalysts are commonly applied in this context to mediate the desired transformations effectively. In particular, the modification of electrode surfaces has proven to be a powerful approach for enhancing selectivity in electrochemical processes. For example, functionalizing carbon-based electrodes with nitrogen-containing groups creates active sites that facilitate the adsorption and transformation of reactive intermediates, improving the overall catalytic activity[24]. Similarly, electrodes modified with conductive polymers, such as polyaniline, not only provide a stable platform for immobilizing catalysts but also allow for precise control over the electrochemical environment, enabling greater selectivity and reactivity[24]. These modifications exemplify how surface engineering of electrodes can direct reactions toward desired products while improving both efficiency and selectivity.
Organic synthesis via divided cells
Electrolysis reactions can be conducted under potentiostatic or galvanostatic conditions (constant potential and constant current, respectively). In potentiostatic electrolysis, a constant potential is maintained using a reference electrode, such as the normal hydrogen electrode (NHE), the saturated calomel electrode (SCE), Hg/HgO or the Ag/AgCl electrode[16]. A high concentration of the starting material ensures mass transport to the electrode, generating a constant charge flow; however, as the substrate is consumed, the current decreases. In addition to the requirement for a reference electrode, another disadvantage is that potentiostats are typically expensive instruments.
On the other hand, while potentiostatic electrolysis typically requires a reference electrode, it is important to note that some systems can operate with only two electrodes (galvanic electrolysis), maintaining a constant potential. In these cases, the potential difference is controlled between the working and counter electrodes, which can technically be considered as potentiostatic control.
In galvanostatic electrolysis, only the anode and cathode are required to perform the reaction under continuous current conditions. In this case, the potential is not controlled, and the substrate with the lowest redox potential is transformed. However, as the starting material is consumed, byproducts can form due to over-electrolysis of products, solvents, or additives. Most electrochemical organic reactions are conducted under galvanostatic conditions, and as a reference electrode is not necessary, simple and inexpensive power sources (DC-Type) can be used, making the systems more practical and feasible for scale-up.
Solvents and electrolytes are responsible for increasing conductivity and reducing the resistance of the medium in electrochemical cells. The most used organic solvents are polar aprotic, such as acetonitrile (MeCN) and dimethyl sulfoxide (DMSO), which need to dissolve and dissociate ionic salts and the organic substrates. Other solvent properties, such as polarity, electrophilicity, nucleophilicity, acidity, basicity, and oxidation/reduction potentials, must be considered since the solvent is exposed to high redox potentials at the electrodes. The use of supporting electrolytes is frequently necessary to maintain charge neutrality in the cell and improve medium conductivity. Electrolytes can include inert (high redox potential) salts, ionic liquids and organic salts, such as n-Bu4NBF4. Another significant advancement involves dual-function electrolytes, which not only support ion transport but also actively engage in the reaction mechanism. For example, certain ionic liquids have been reported to stabilize radical intermediates, thereby enhancing selectivity and preventing unwanted side reactions[25]. This dual role transforms the electrolyte from a passive component into an active player in optimizing the overall efficiency and selectivity of the electrosynthesis process. The design and selection of electrolytes must, therefore, consider not only their inertness and conductivity but also their potential interaction with reactive species and substrates. By tailoring these interactions, it is possible to fine-tune reaction conditions, optimize energy demands, and improve yields, making the electrolyte a crucial element in the development of innovative electrosynthesis methodologies[25].
Additionally, the formal redox potential determines the equilibrium between the oxidized and reduced forms of a chemical species and is influenced by variables such as solvents, temperatures, and supporting electrolytes, and its control is essential to avoid side reactions and ensure selectivity in electrosynthesis[26]. Overpotential, defined as the difference between the applied potential and the formal potential, reflects system resistance and affects charge transfer kinetics and mass transport limitations, directly impacting process efficiency[27]. Similarly, Faradaic efficiency, which measures the conversion of electrical current into desired products, is fundamental for process optimization, minimizing energy losses in non-productive reactions[28]. Notably, in specific applications such as drug production, controlling these variables is crucial, especially to achieve enantioselectivity. This enantioselectivity can be enhanced using modified electrodes or chiral mediators, favoring the formation of a specific enantiomer and ensuring the desired selectivity[29].
Additionally, the control of selectivity in electrochemical transformations can be achieved through innovative approaches, such as the redox-tag approach[30], the cation-pool method[31-34], and bio-electrochemistry[7,16].
Selection of membranes
The selection of membranes significantly influences system performance, efficiency, and longevity. Perfluorosulfonic acid (PFSA)-based membranes, such as Nafion, are the benchmark due to their high proton conductivity (~0.07-0.08 S/cm, Siemens per centimeter), chemical stability, and mechanical durability, though their high cost and reliance on fluorinated polymers raise economic and environmental concerns. Alternative materials, such as sulfonated poly(ether-ether-ketone) (SPEEK) and sulfonated poly(aryl ether sulfone) (SPAES), offer lower costs and similar ionic conductivity in hydrated conditions but face limitations in durability and water swelling. Hybrid membranes, incorporating nanofillers such as silica or metal-organic frameworks (MOFs), enhance mechanical and thermal properties while boosting conductivity, with examples such as silica-blended SPEEK achieving 0.22 S/cm at 70 °C. However, challenges remain in processing complexity and long-term compatibility. Advanced composite membranes reinforced with inorganic fillers or crosslinked structures improve conductivity and stability at elevated temperatures but require balancing performance, durability, and cost. Continued innovation is essential to develop sustainable, cost-effective alternatives that rival or surpass Nafion’s performance while addressing scalability and environmental impact[14,35].
Furthermore, operational stability plays a crucial role in the economic efficiency of electrosynthetic reactions, complementing advances in selectivity and efficiency. Factors such as the durability of electrodes and membranes, resistance to contamination by byproducts, and the maintenance of Faradaic efficiency over time are critical for reducing operational costs. In large-scale applications, the stability of cell components directly influences productivity and economic competitiveness, making its optimization indispensable[24,36,37]. These systems, despite presenting challenges such as scalability for large-scale production and the implementation of selective membrane filtration technologies, hold immense promise. Divided cells have the potential to drive the development of sustainable and highly efficient chemical processes, addressing key limitations in modern electrosynthesis.
Scope of this review
This review discusses the principles, advantages, and challenges of using divided cells in electrosynthesis reactions. The fundamentals of electrosynthesis and the types of cells applied for these reactions will be introduced, emphasizing the benefits and drawbacks of divided cells. Recent advances in employing divided cells for organic compound synthesis are highlighted, focusing on their potential for sustainable and efficient chemical processes. Factors influencing the performance of divided cells, such as electrode materials, membrane types, and operating conditions, are considered. This review concludes by analyzing the challenges associated with divided cell electrosynthesis and provides future perspectives.
DIRECT ELECTROLYSIS
In direct electrolysis, ET occurs directly between the organic substrate and the electrode, characterizing a heterogeneous process. The reaction mechanism involves the interaction of species at the electrode surface, where their transport can result from migration (for charged species under the influence of an electric field), diffusion (due to concentration gradients), or convection (caused by fluid motion). Neutral species do not undergo migration, as their transport is primarily governed by diffusion. Migration, on the other hand, predominantly influences charged species, including ions from the supporting electrolyte and ionic reactants or intermediates, ensuring electrical conductivity in the system[38,39].
The oxidative cross-coupling of aromatic carbon-hydrogen (C–H)/C–H bonds has gained significant interest among synthetic chemists, as it avoids the need for pre-functionalization of starting materials and provides a direct, step-efficient method to form a single C–C bond between two aromatic rings[40,41]. Morofuji et al. developed an efficient and regioselective method for the cross-coupling of C(sp2)–H/C(sp2)–H bonds in two deactivated aromatic compounds using the methodology of radical-cations pools in electrochemical systems [Scheme 1][42]. A divided cell was applied with a carbon felt (CF) electrode at the anode and platinum at the cathode.
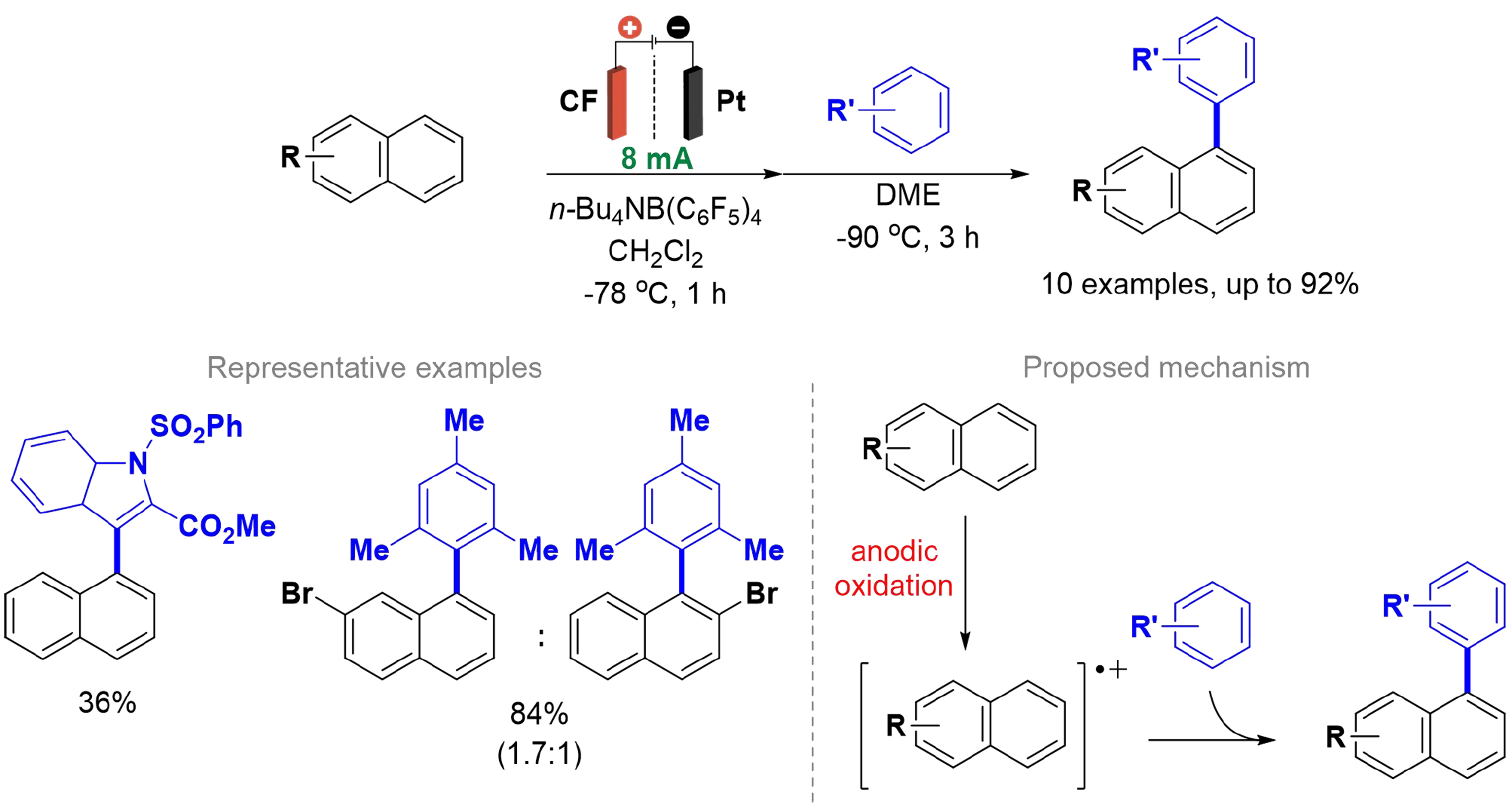
Scheme 1. Cross-coupling of C(sp2)–H/C(sp2)–H bonds in two deactivated aromatic compounds.
The radical cations pools are intended to prevent the formation of homo-coupling reactions. It was observed and confirmed that only 1-substituted naphthalene derivatives were obtained, and the corresponding 2-substituted product was not detected, demonstrating the total regioselectivity of the reaction. The authors performed density functional theory (DFT) calculations that suggested a favored reaction at the 1-position. This was consistent with the high regioselectivity reported for the reaction of naphthalene radical cations with other nucleophiles[43]. Furthermore, the bromo-substituted naphthalene already had the ortho-position product formed in lower amounts, which agreed with the DFT calculations. After undergoing anodic oxidation, naphthalene radical cations, or their derivatives, form at low temperatures. Subsequently, they react with the second aromatic compound to yield the respective cross-coupling product. The method proved to be advantageous as it is selective for performing C–H/C–H cross-coupling of aromatic compounds and does not require the use of metal complexes or chemical oxidants.
The nucleophilic aromatic substitution of hydrogen (SNH reactions)[44] through electrochemical methodologies offers a pathway for the direct functionalization of C(sp2)–H bonds in aromatic compounds without the use of stoichiometric amounts of oxidants. Successful applications of electrochemical SNH reactions for the functionalization of nitroarenes by the addition of C-, N-, O-, and P-based nucleophiles are described[45]. Additionally, Chupakhin et al. demonstrated the modification of aza-aromatic compounds
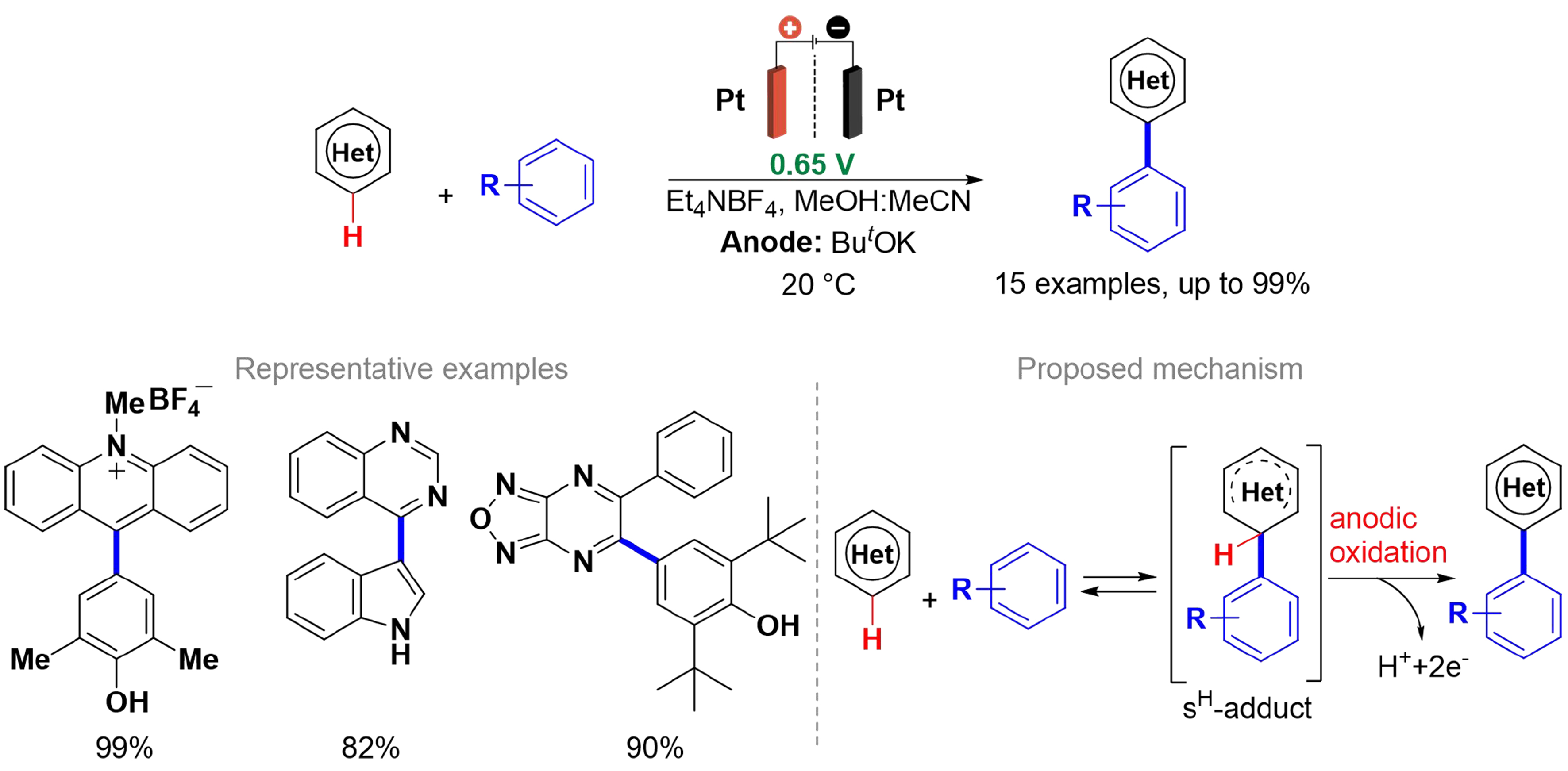
Scheme 2. C(sp2)–H functionalization of aza-aromatic compounds.
The acridinium cation and aza-aromatic compounds undergo efficient arylation and heteroarylation with phenols, indoles, pyrroles, and anilines. The method utilizes electrical energy without relying on external oxidants and eliminates the need for intermediate isolation and purification. SNH reactions proceed in two stages: during the formation of the new C–C or C–X bond, the reversible addition of the nucleophilic species to the aromatic ring forms an intermediate known as the σH-adduct. The anodic oxidation of the σH-adduct (elimination of H+ + 2e-) yields the coupling product, with potential control being crucial for the success of these reactions.
In 2023, Sun et al. investigated the regioselectivity of N-heteroarene electrocarboxylations, emphasizing the distinctions between divided and undivided electrochemical cells [Scheme 3][47]. The authors highlight the importance of site-selective C–H functionalization in organic synthesis, particularly through the carboxylation of N-heteroarenes using carbon dioxide. The study demonstrates that the type of electrolytic cell applied can significantly influence the regioselectivity of the carboxylation process. Specifically, C5 carboxylation is favored in divided cells, while C4 carboxylation occurs in undivided cells. The mechanistic study suggests that C5 carboxylation on a pyridine ring begins with a single-electron reduction, forming a radical anion. This radical then reacts with CO2 at the C5 position due to higher electron density. The intermediate product is further reduced, followed by oxidative rearomatization, resulting in the carboxylated product. However, DFT calculations show that this nucleophilic addition of CO2 is reversible and can occur at either the C5 or C4 positions, with regioselectivity depending on the conditions. In an undivided cell, the hydrogen acceptor generated at the anode favors C4 carboxylation via hydrogen transfer or proton-coupled ET[48].
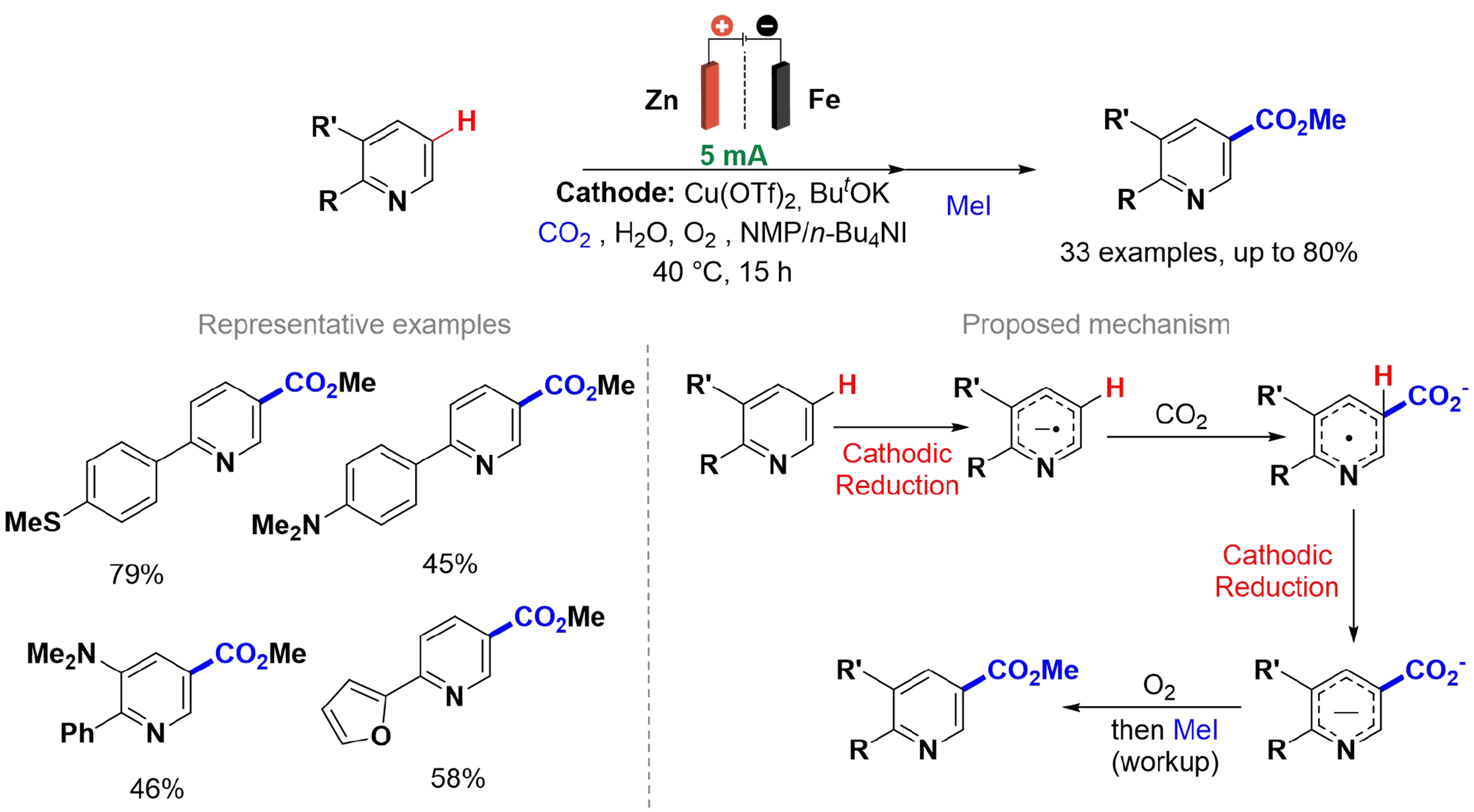
Scheme 3. C5-carboxylation via electro-reductive activation in a divided cell.
Fluorinated substrates, particularly molecules containing the trifluoromethyl group, are of significant pharmaceutical interest. To obtain these compounds, trifluoromethanesulfinates act as a source of trifluoromethyl radicals under oxidative conditions, such as in the presence of tert-butyl hydroperoxide (TBHP), and are utilized for the trifluoromethylation of aromatic compounds and heterocyclic substrates[49,50]. However, this process typically requires a large excess of oxidant and sulfinate, and low yields are frequently observed. O’Brien et al. described a C–H functionalization of heteroarenes using electrochemical initiation of radicals. The reactions were conducted in a divided cell setup with a carbon cloth anode as the working electrode and a carbon cathode in an auxiliary compartment, fitted with a polytetrafluoroethylene (PTFE) frit [Scheme 4][51]. Constant current electrolysis provides direct control over the oxidation rate of the sulfinate and enables the controlled generation of radicals. In the reaction mechanism, the rate-determining step involves ET from the anion to form the sulfinate radical, followed by rapid cleavage to produce the fluoroalkyl radical, which then couples with the substrate to generate the desired product[52].
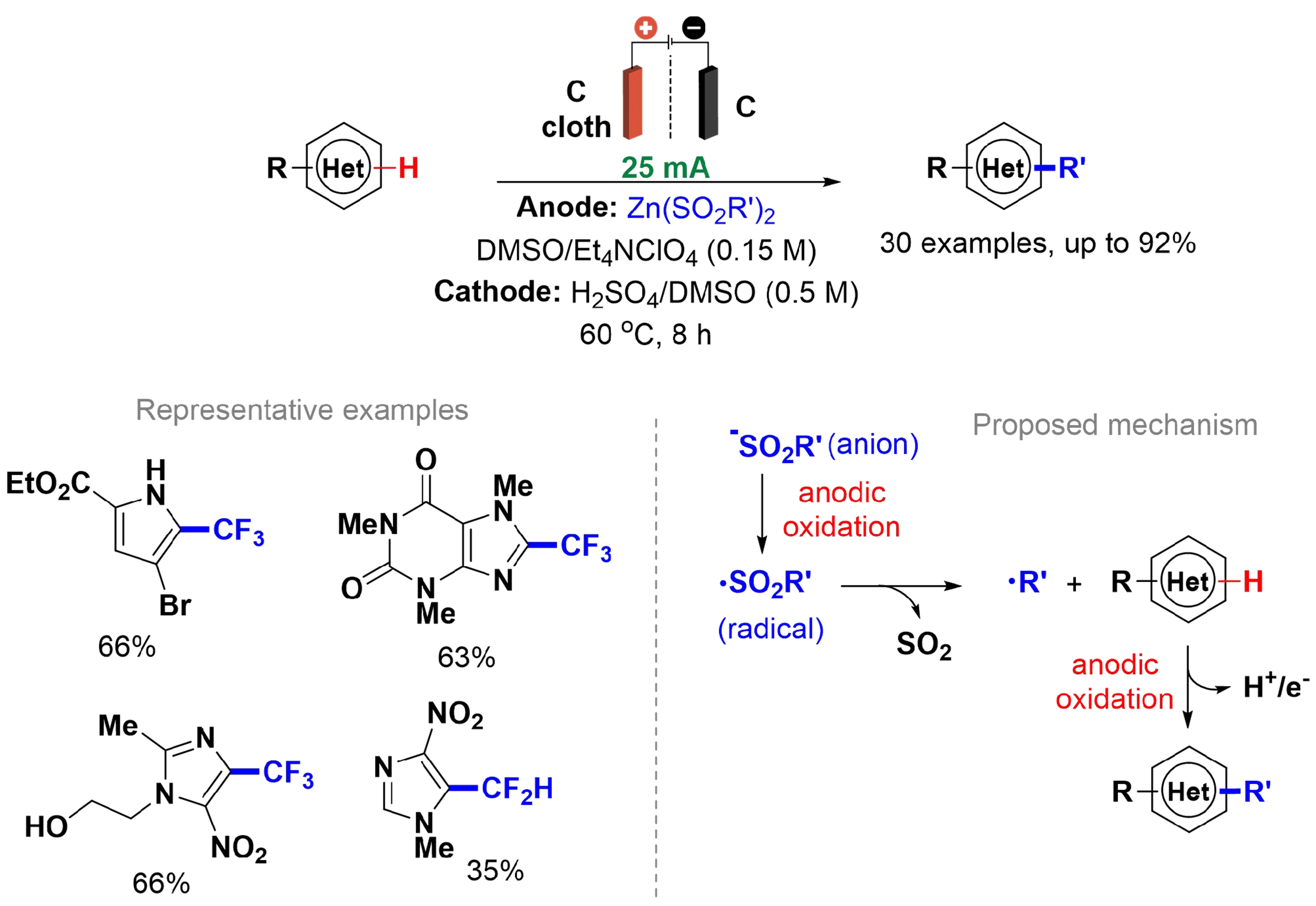
Scheme 4. C(sp2)–H functionalization of heterocyclic substrates by electrochemically initiated sulfinate-derived radicals.
Since several 4-aminonaphthalen-1-ols have been reported as inhibitors of enzymes and protein-protein interactions[53-57], 4-aminonaphthalen-1-ols substituted at positions 2- and 3- hold potential for application as bioactive molecules. In light of this, Kise et al. proposed their synthesis involving the electro-reductive coupling of phthalimides with α,β-unsaturated carbonyl compounds in the presence of trimethylsilyl chloride (TMSCl) [Scheme 5][58]. The electrolysis was carried out in a divided cell, equipped with a ceramic cylindrical diaphragm and platinum electrodes, using Et4NOTs as the supporting electrolyte. The authors applied DFT calculations to suggest that the substituent R on the nitrogen of the phthalimides affects product formation. Bulky substituents favor O-protonation, leading to 3-substituted products, while less bulky groups promote faster N-protonation, resulting in substitution at the 2-position. The reduced phthalimide couples with the carbonyl compound to form an enolate, which undergoes O-silylation to yield the silyl ketene acetal[59]. The acid-catalyzed rearrangement then occurs through intramolecular electrophilic addition, ring expansion, desilylation, protonation, and dehydration, resulting in the rearranged products.
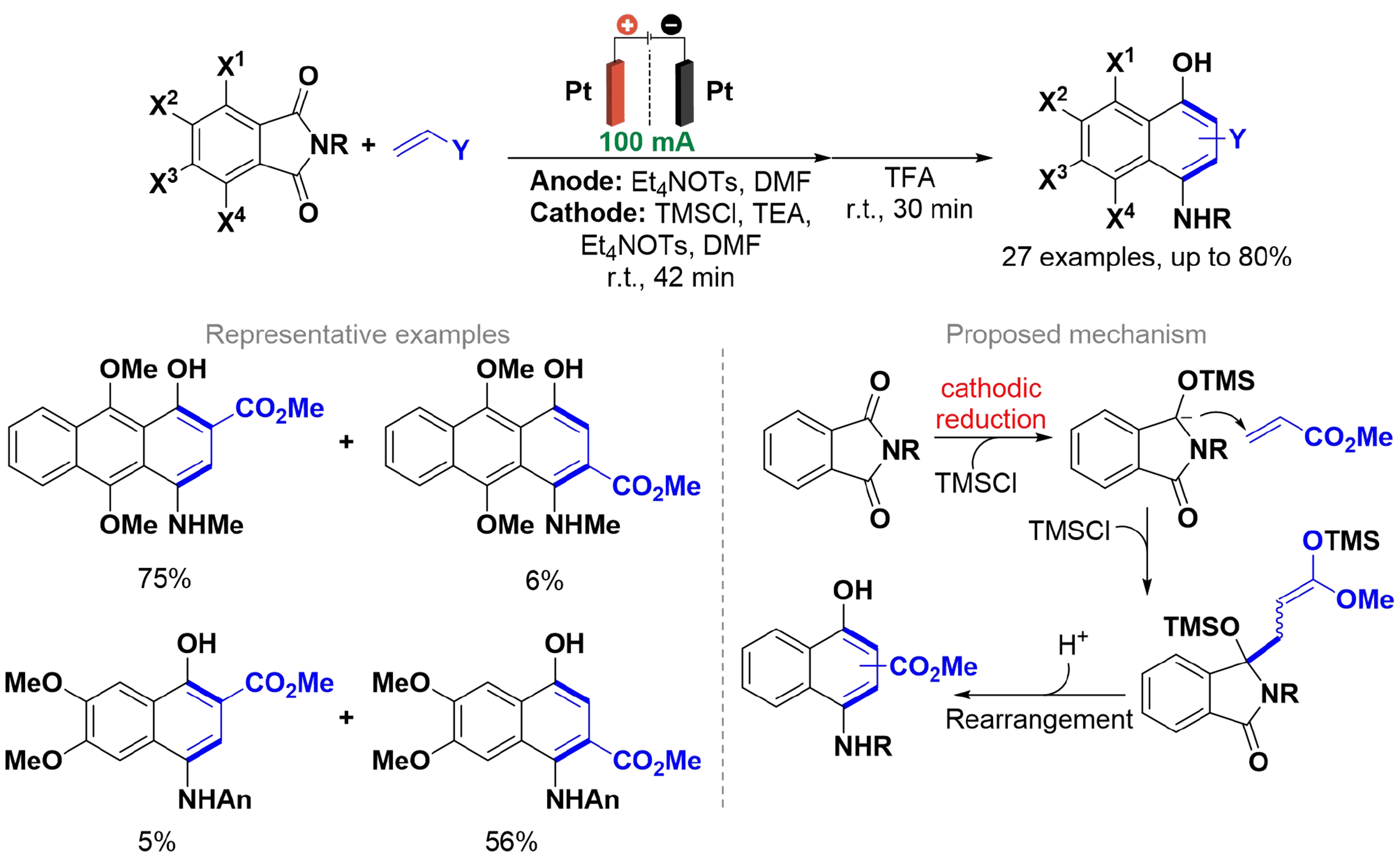
Scheme 5. Electro-reductive coupling of phthalimides with α,β-unsaturated carbonyl compounds and acid-catalyzed rearrangement.
Primary aromatic amines with specific functional groups are crucial for producing various organic materials, including dyes, pigments, medicinal compounds, and agrochemicals[60,61]. Several synthetic methods have been developed from corresponding aromatic compounds, with the most common involving nitration followed by the reduction of nitro groups. However, this process requires aggressive chemicals, and frequently, transition metals[62]. Morofuji et al. reported an approach using an electrochemical oxidation of aromatic compounds in the presence of equimolar pyridine and piperidine, in several reactions to produce the primary aromatic amines [Scheme 6][63]. A divided cell with a CF electrode at the anode and platinum at the cathode was used. The choice of pyridine as the nitrogen source was based on its ability to enable the selective oxidation of aromatic compounds, by the formation of N-arylpyridinium ions preventing overoxidation, making it an option for various synthetic applications[64,65]. Anisole was selected as the basic substrate due to its lower oxidation potential compared to pyridine. The method yielded products for both substituted anisoles and π-extended aromatic compounds, with the regioselectivity predicted by DFT calculations. The reaction begins with the one-electron oxidation of anisole, forming a radical cation, followed by the attack of pyridine, leading to the N-arylpyridinium ion. The reaction with piperidine occurs via addition to the 2-position of the N-arylpyridinium ion, followed by ring opening and imine hydrolysis.
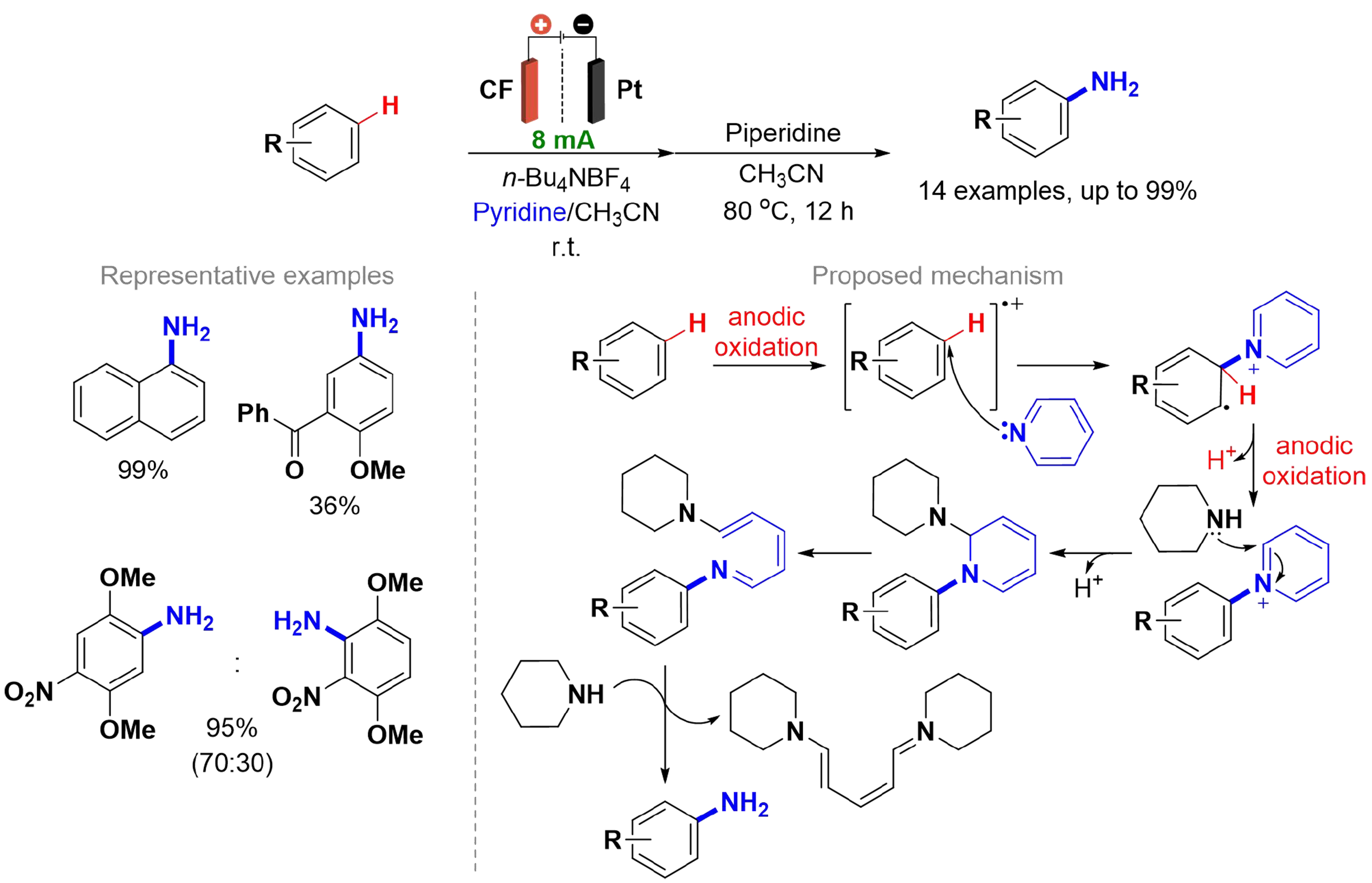
Scheme 6. Electrochemical oxidation to produce primary aromatic amines.
Benzoxazoles and benzothiazoles are key cores of natural products and bioactive compounds, including antiviral and antitumor agents[66,67]. Their synthesis via electrochemical amination of C–H bonds was developed by Morofuji et al. using a divided cell with a CF anode, a platinum cathode, and a porous glass membrane [Scheme 7][68]. The primary motivation behind the development of this methodology by the authors was to eliminate the need for transition metals, which were typically used to synthesize these compounds, despite their involvement in the preparation of the starting materials[69,70].
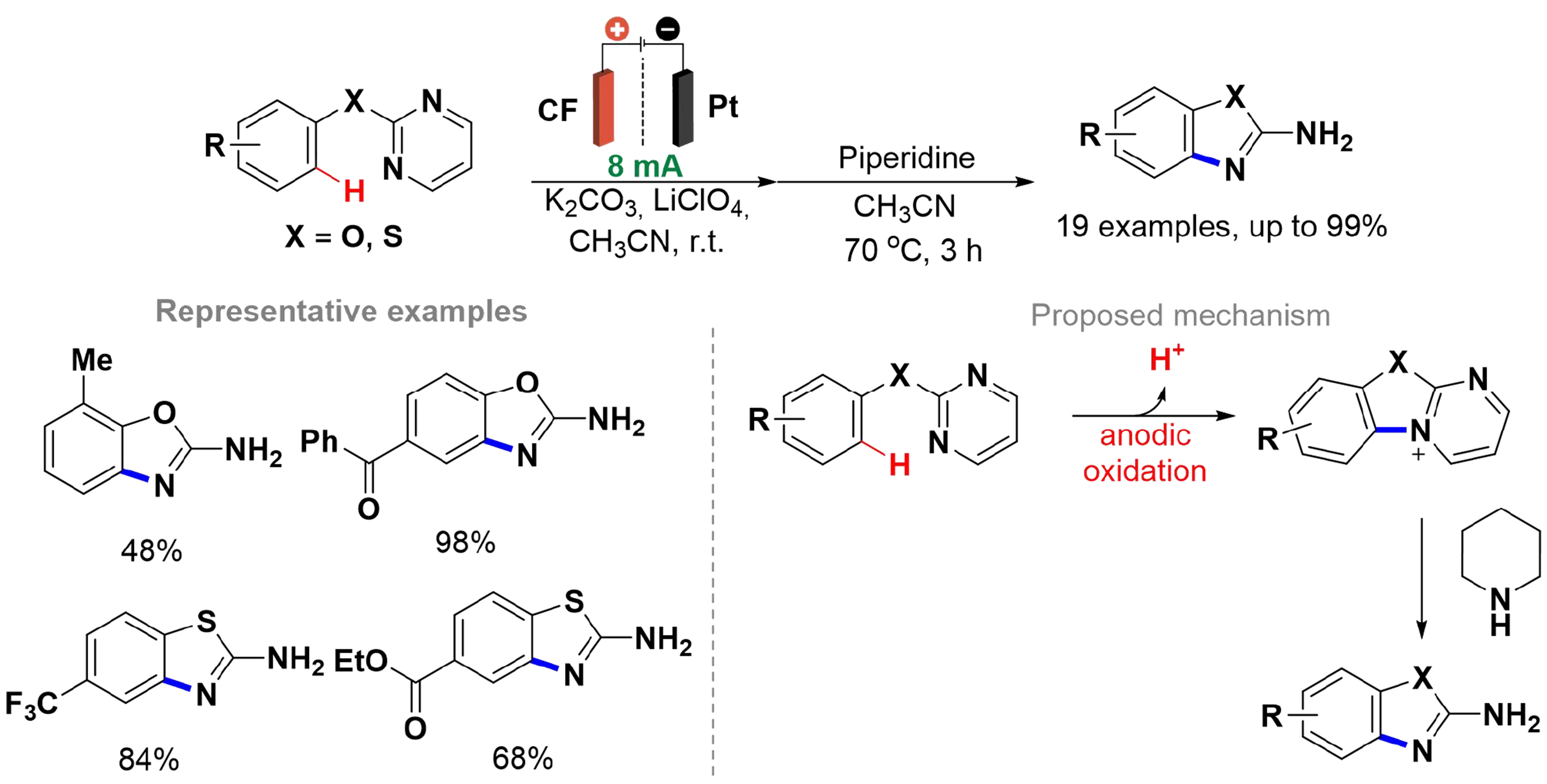
Scheme 7. Synthesis of benzoxazoles and benzothiazoles by electrochemical intramolecular C–H amination.
An aspect of this approach is that the amination reaction of 2-pyrimidinyloxybenzenes and 2-pyrimidinylthiobenzenes occur intramolecularly. The method included halogenated substituents, and was compatible with carbonyl and carboxyl groups, achieving yields of up to 99%. In the proposed mechanism [Scheme 7], the process began with a single-electron oxidation followed by an intramolecular attack by the nitrogen atom of the pyrimidine ring. This was succeeded by another single-electron oxidation and proton removal, leading to the formation of a cyclic cationic intermediate. Finally, the addition of piperidine resulted in the generation of the desired products.
In 2017, Wesenberg et al. reported an electrochemical synthesis of 1,4-benzoxazin-3-one derivatives from phenoxyacetates using pyridine as the nitrogen source, without the use of transition metal catalysts
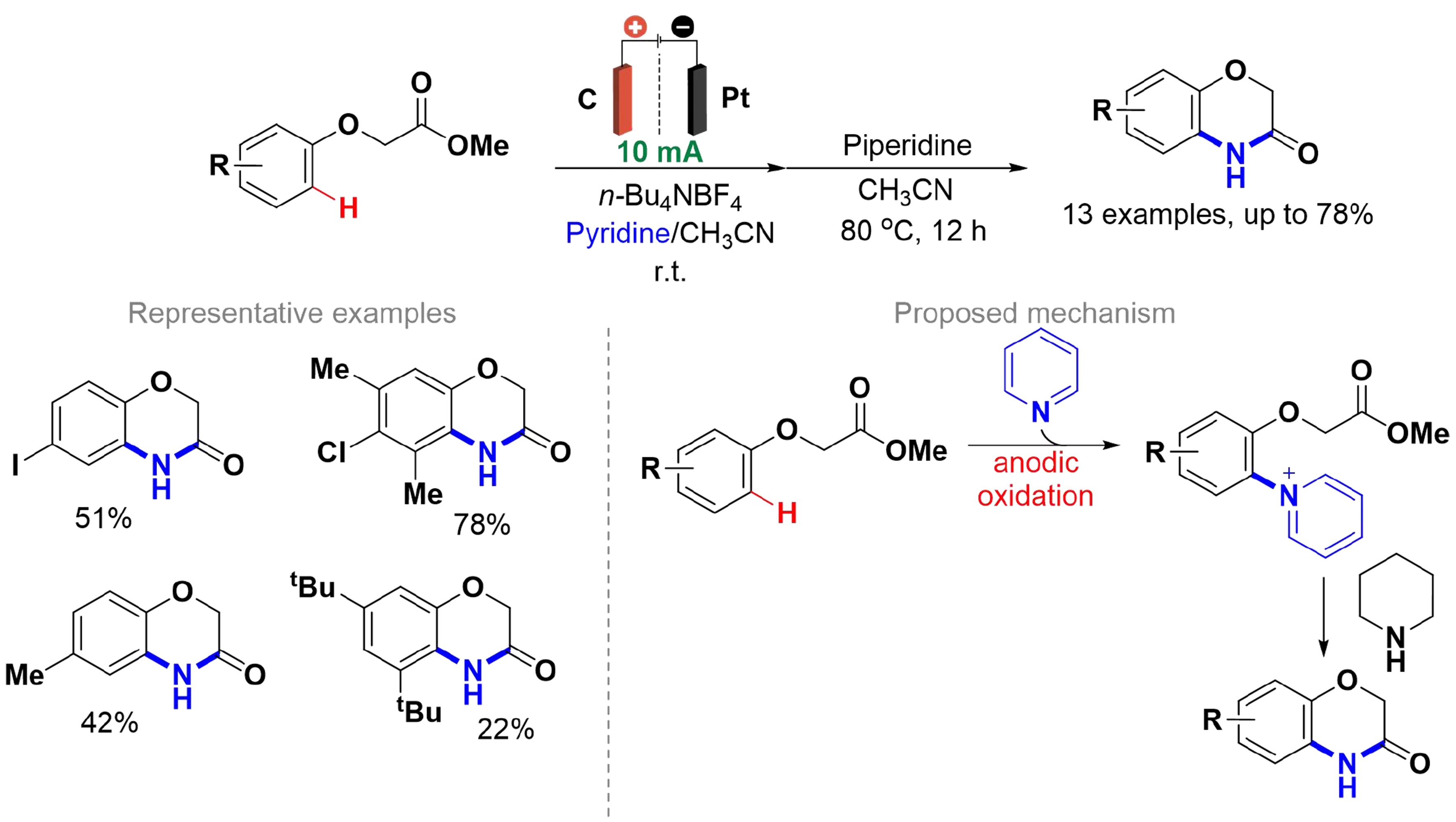
Scheme 8. Electrosynthesis of 1,4-benzoxazin-3-one derivatives using pyridine as the nitrogen source.
The amide bond is the essential linkage that forms the backbone of proteins, peptides, and many other biologically significant compounds. Furthermore, they are present in agrochemicals with herbicidal and antifungal activities and in marketed pharmaceuticals. Kononov et al. developed a sustainable electrochemical approach for the direct amidation of aromatic C–H bonds [Scheme 9][73] eliminating the need for sulfuric acid commonly used in previous protocols[74]. The method employs a divided cylindrical cell using a ceramic plate with a pore size of 900 nm as a membrane and nitrile solvents as sources of amide and amine groups, with Pt/Pt electrodes, allowing the synthesis of 60 N-phenylamides and 12 benzoxazoles under mild, metal- and oxidant-free conditions.
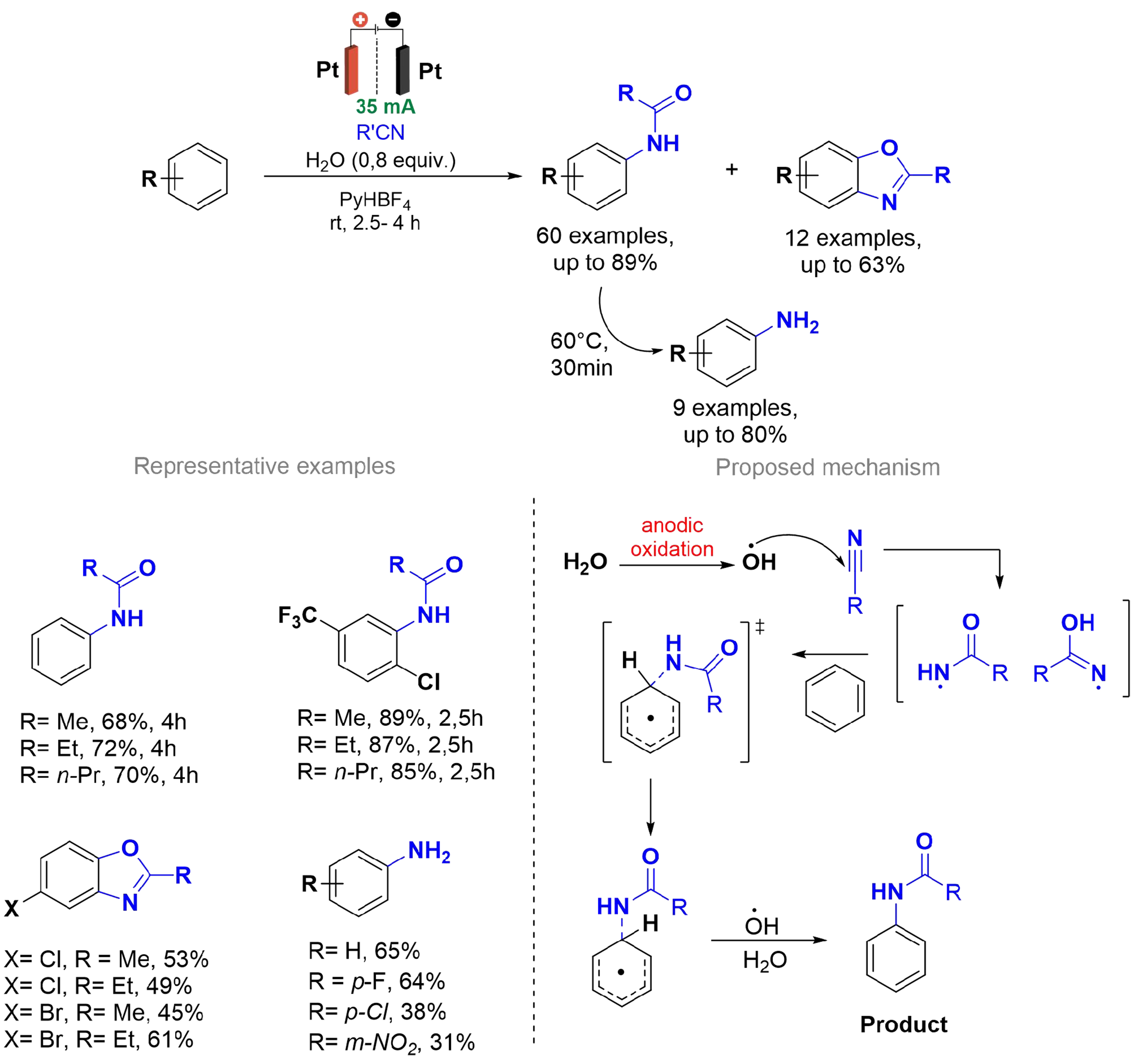
Scheme 9. Ritter-type electro-oxidative aromatic C–H amidation of arenes.
Mechanistic studies revealed that the reaction begins with the formation of hydroxyl radicals from residual water at the anode, which then reacts with nitriles to generate amide radicals that are subsequently added to the arenes. This methodology achieved high yields (up to 89%) and enhanced selectivity, demonstrating its applicability to gram-scale synthesis and paired electrosynthesis. This advancement underscores the potential of electrochemistry for C–H functionalization reactions as a sustainable alternative to more traditional methodologies.
Aryl iodides are important building blocks, as they react in homo- and cross-couplings, in reactions catalyzed by transition metals[75,76] and are precursors of organometallic reagents such as organolithium and Grignard reagents[77]. Traditional methods for obtaining these compounds are based on electrophilic aromatic substitution with iodine[78,79] or hypervalent iodine[80], which are methods that require the use of an excess of halogens and oxidants. In 2018, Sano et al. reported the iodination of N-(8-quinolinyl)benzamide derivatives using I2 under electrochemical conditions, conducted in an H-type divided cell equipped with two platinum electrodes and an anion exchange membrane at a temperature of 90 °C [Scheme 10][81]. Initially, ortho-substituted compounds were evaluated under electrochemical and non-electrochemical conditions, yielding similar results. For meta- and para-substituted benzamides, both mono- and diiodinated products were formed, with higher yields of the diiodinated product under electrochemical conditions, indicating that anodic oxidation effectively promotes iodination at sterically hindered positions. The Pd(II) catalyst reacts with benzamide to form a chelate by breaking the C–H bond, which then reacts with I+ generated from the anodic oxidation of I2. This method enables the iodination of benzamide derivatives with both electron-withdrawing (EWG) and electron-donating groups (EDG) in non-electrochemical and electrochemical conditions, the latter enhancing diiodination in meta-substituted benzamides.
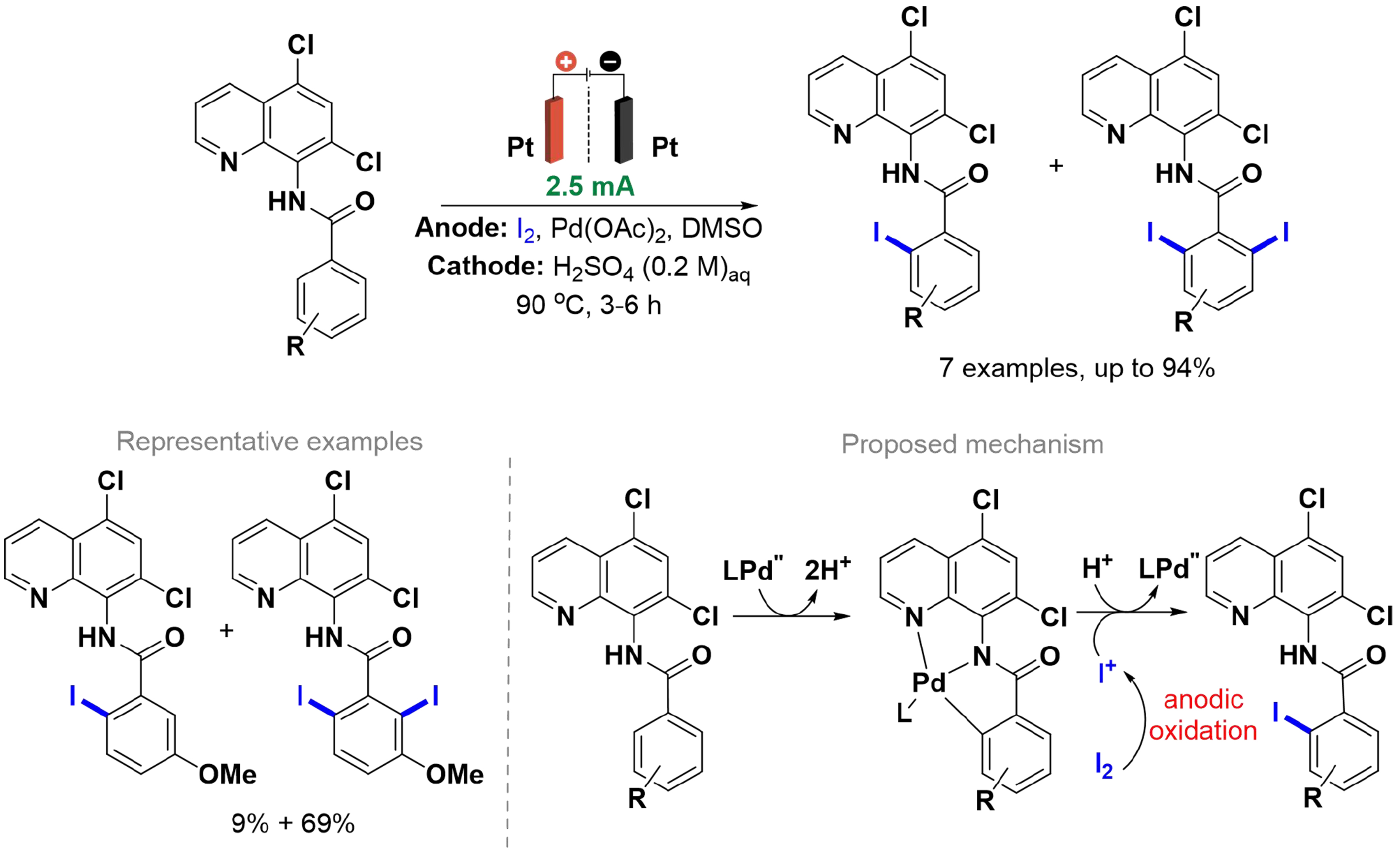
Scheme 10. Pd-catalyzed C(sp2)–H iodination of N-(8-quinolinyl)benzamide derivatives.
Möckel et al. reported an electrochemical method for iodinating aromatic compounds employing the trimethylsilyl group to direct the iododesilylation reaction [Scheme 11][82]. In addition to enhancing selectivity, the use of a directing group increased reactivity and reduced reaction time. However, this approach presented a lower atom economy compared to the method described in Scheme 10. Potassium iodide (KI) was utilized as the iodine source, which eliminated the need for a supporting electrolyte in the anodic compartment. The reactions were carried out at room temperature using an H-type divided cell, platinum electrodes, and a solvent mixture of MeCN and methanol (9:1), with sulfuric acid acting as the supporting electrolyte in the cathodic compartment. A design of experiments (DoE) was applied to develop an optimal model for o-, m-, and p-substituted arenes[83,84], yielding excellent results for both EDG and EWG. The methodology showed compatibility with various functional groups[85], including aromatic halides, nitro, nitrile, amides, esters, and ketones. However, it was unsuitable for heteroaromatic compounds, alcohols, primary amines, alkenes, and alkynes. Notably, this method facilitated the synthesis of isotopically labeled molecules, exemplified by the successful synthesis of Boc-protected 3-iodobenzylguanidine, which was obtained in 88% yield without purification.
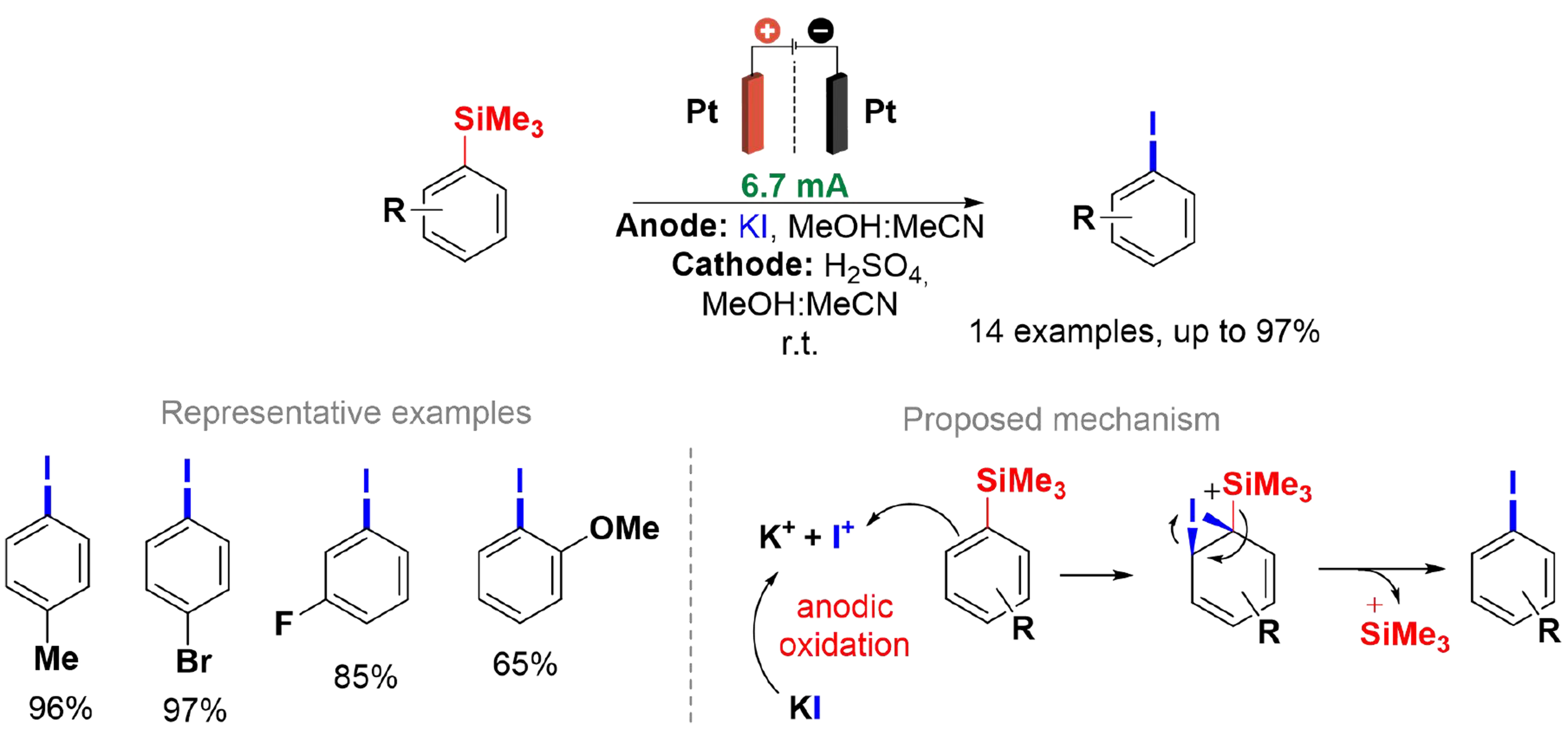
Scheme 11. Anodic iododesilylation reaction.
Although some methodologies use a platinum working electrode[82,86,87], Yan et al. report that the release of Pt ions into the reaction medium can affect the regioselectivity of aromatic electrophilic substitution (SEAr) [Scheme 12][88]. The studies were conducted using an H-type divided cell separated by a 4G glass sand core, and constant current electrolysis was conducted using a Pt cathode and anode. Glassy carbon, Pt and graphite electrodes were applied, and the regioselective iodination of anisole was evaluated, which provided 4-iodoanisole in 97%, 16% and 60% yield, respectively. Contamination by Pt ions was confirmed by inductively coupled plasma mass spectrometry (ICP-MS) analysis, detecting traces of Pt (~1 μg/mL) in the solution, which may justify the low yield of the product when the Pt anode was used. Therefore, the iodination of aromatic substrates was performed using a glassy carbon anode, providing excellent yields. The cathodic reduction of H+ to H2 occurs concurrently with the anodic oxidation of I2 at the glassy carbon anode in this process, generating the I+ species that reacts with aromatic substrates, resulting in selective iodination at the para position.
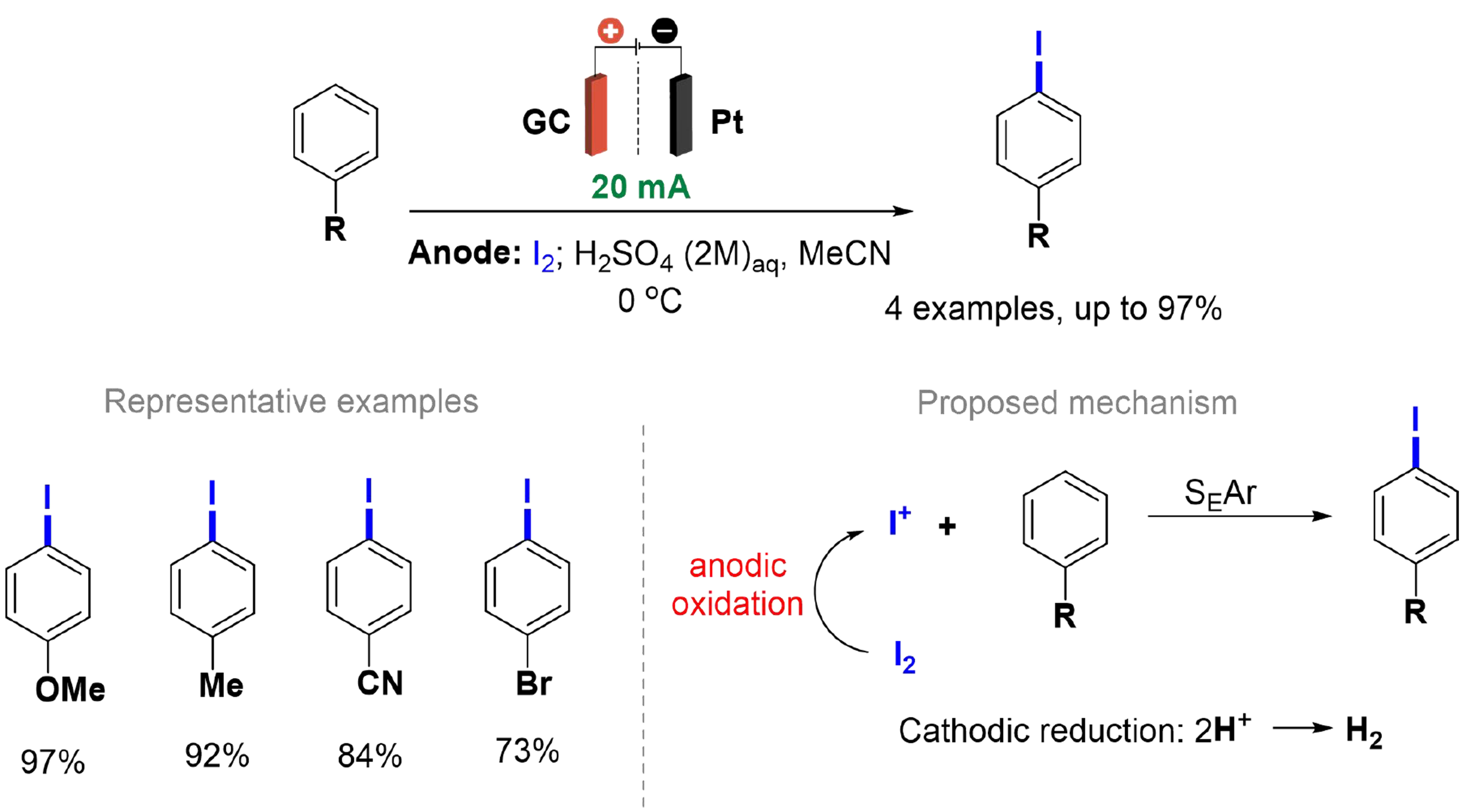
Scheme 12. Regioselective iodination of aromatic substrates.
Aryl bromides, similar to aryl iodides, are essential intermediates in the synthesis of organometallic reagents and in coupling reactions[89,90]. Consequently, C–H bromination through electrochemical methods is highly valuable, making it significant for synthetic applications. Yang et al. developed a palladium-catalyzed method for the C(sp2)–H bromination of benzamide derivatives [Scheme 13][91]. Applying an H-type divided cell equipped with an anion-exchange membrane, a platinum plate cathode, and a reticulated vitreous carbon (RVC) anode, the bromination of benzamide was performed under constant current electrolysis in the presence of NH4Br, which acts as both an electrolyte and a source of Br- ions. The proposed mechanism involves palladium coordinating with the benzamide via oxidative addition to form a Pd(II) complex, which then reacts with Br3- or Br2 generated by anodic oxidation to produce high-valent Pd(III) or Pd(IV) species. Reductive elimination regenerates a Pd(II) complex that exchanges ligands to yield the final product. The divided electrochemical cell with an anion exchange membrane is essential, as the oxidation of NH4Br provides a source of bromine while preventing parallel reduction reactions.
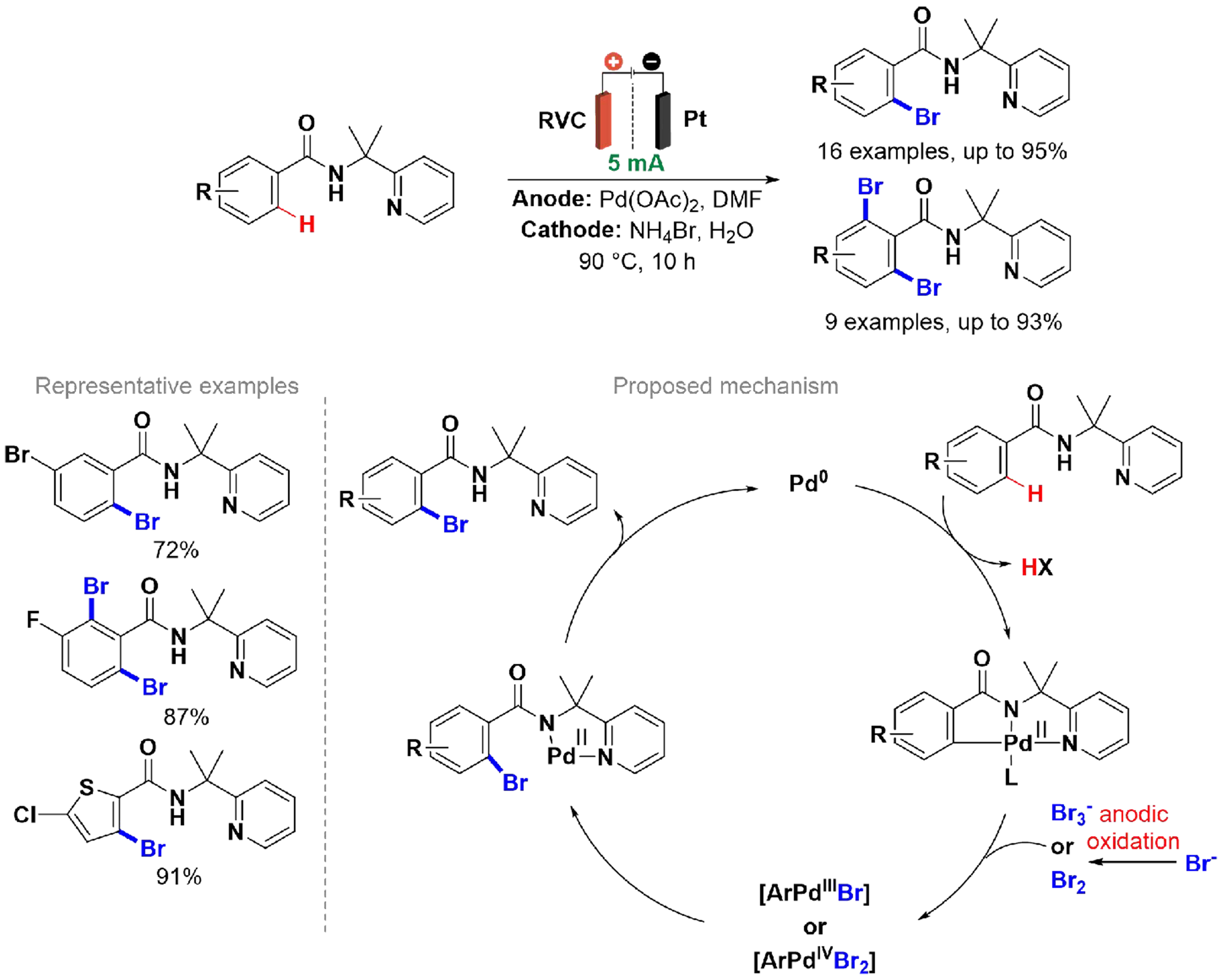
Scheme 13. Pd-catalyzed C(sp2)–H bromination of benzamide derivatives.
An approach to maximize both Faradaic and synthetic efficiency, along with atom economy in electrolysis reactions, is to couple the reduction and oxidation reactions that occur simultaneously at the electrodes to produce the desired products[92,93]. Benzyl halides are widely utilized building blocks in the fine chemical industry, and a green approach to obtain these organic halides is through electrophotocatalytic radical bromination. This electrooxidation reaction can be paired with the electrochemical reductive coupling of carbonyl compounds, enabling the synthesis of vicinal 1,2-diols without the need for sacrificial electrodes. Chen et al. developed a photoelectrochemical approach for the benzylic bromination of various alkylbenzenes paired with the reductive pinacol coupling of carbonyl compounds [Scheme 14][94].
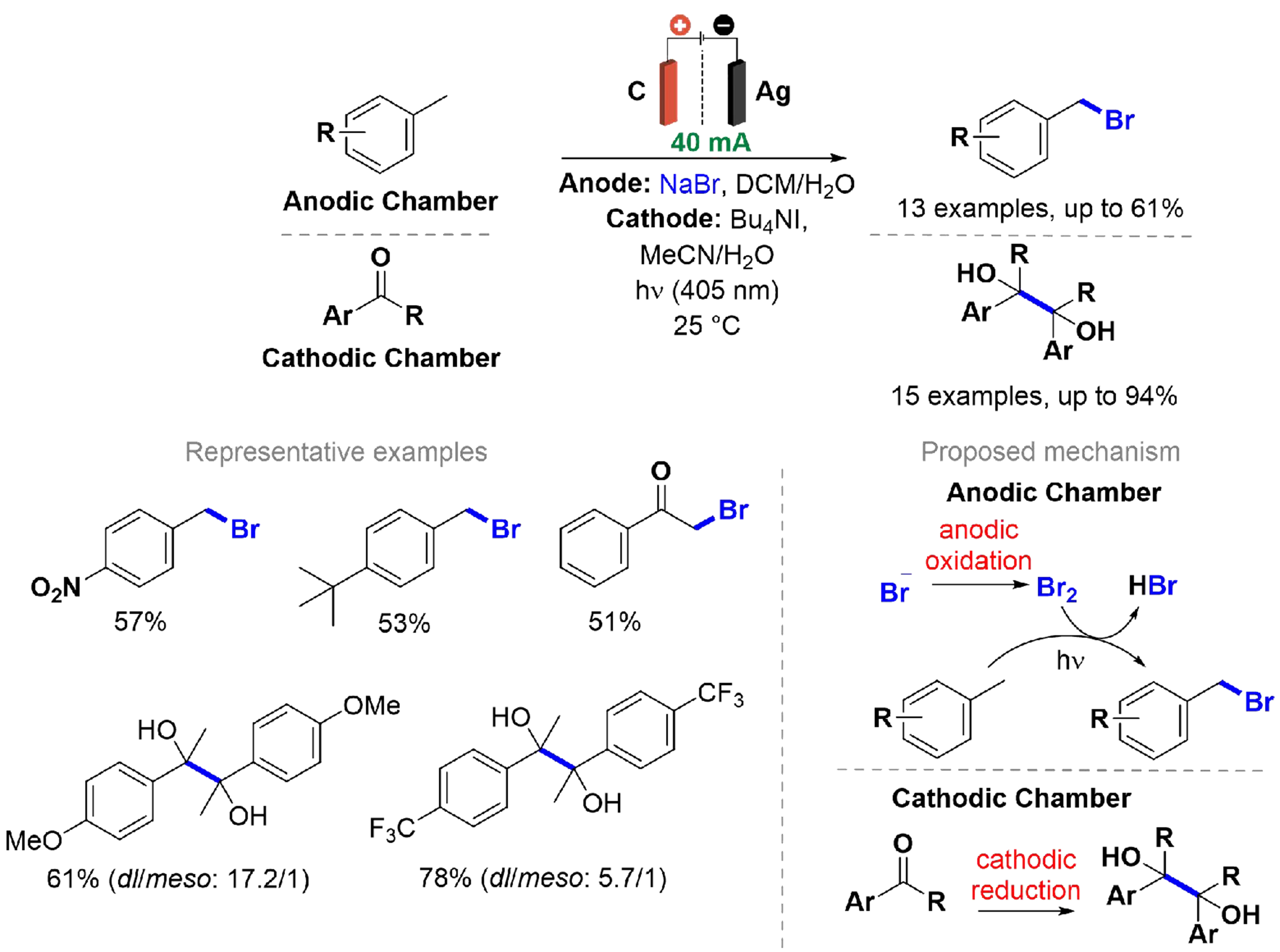
Scheme 14. Electrophotocatalytic benzylic bromination of alkylbenzenes paired with reductive pinacol coupling of carbonyl compounds.
The reactions were carried out in an H-type divided cell, employing distinct cathodic and anodic electrolyte solutions separated by a Nafion 117 membrane. The reaction mixture was irradiated with 405 nm blue light (20 W) at 25 °C under a constant current of 40 mA. Carbon rod and silver flake electrodes were used as the anode and cathode, respectively. All reactions were conducted on a gram scale, and their application to various substrates, including EDG and EWG, resulted in moderate-to-good product yields with high Faradaic efficiencies. Voltammetry analyses indicated that the presence of water in the cathodic compartment could accelerate the reduction of carbonyl compounds, generating a radical intermediate that facilitates dimerization. Nevertheless, a drawback of using a divided cell is that only a portion of the applied voltage drives the electrochemical reaction, with the remainder compensating for Ohmic drops. These losses stem from the low conductivity of the organic electrolyte, the compartmental separation imposed by the Nafion membrane, and mass transfer limitations.
A key intermediate for the synthesis of Ledipasvir, an NS5A inhibitor used in the treatment of hepatitis C, can be obtained through the double dehalogenation of a dibromo-spirocyclopropane-proline derivative. Electrochemical dehalogenation is already a promising approach; however, the methodologies described employ lead or mercury as the cathode[95-100], which results in metal salt contamination in the product, making them incompatible with the production of pharmaceutical intermediates. Gütz et al. developed an efficient electrochemical dehalogenation protocol utilizing leaded bronze as the cathode [Scheme 15][101]. The electrolysis was conducted using a divided cell, featuring a porous ceramic separator and a platinum grid anode. The electrolyte used consisted of dimethylformamide (DMF) and [Et3NMe]O3SOMe. The dibrominated substrate was reduced at the cathode, while methanol was oxidized at the anode as a sacrificial compound. A leaded bronze cathode provided high selectivity, reactivity, and stability. This method permits clean conversion to a single product and enables recycling of the solvent and supporting electrolytes. A flow electrolysis device, separated by a Nafion membrane, gives precise control over reaction conditions and enhances scalability. Key factors influencing the process include current density and flow rate, with optimal dehalogenation conditions being 20 F, 15 mA/cm2, and 0.6 mL/min.
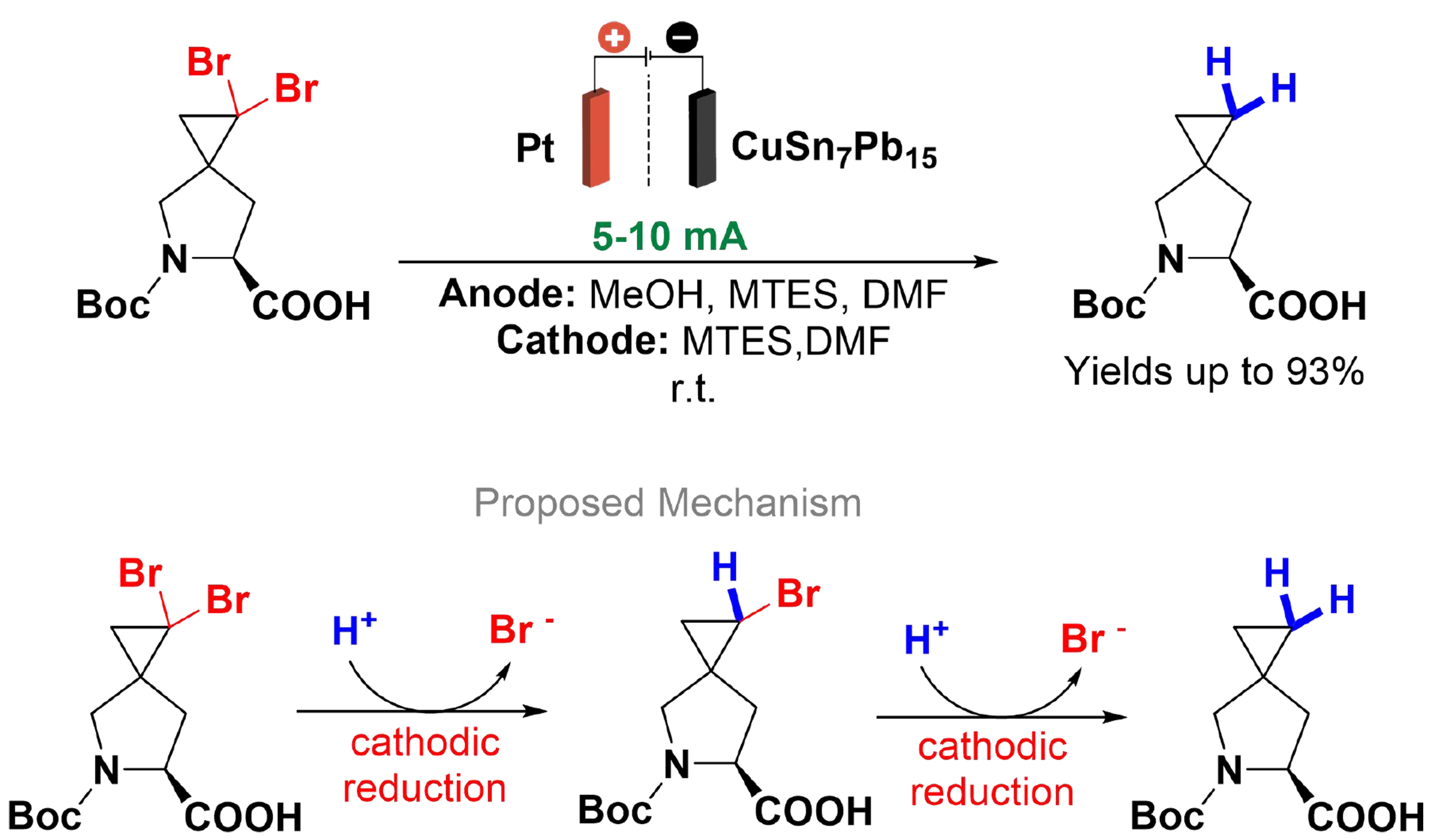
Scheme 15. Electrochemical dehalogenation of a spirocyclopropane-proline derivative.
Chloroarenes are an important scaffold, found in pharmaceuticals and natural products, and are widely used in various organic reactions[102,103]. Their synthesis employed transition metal catalysis and external oxidizing agents. Konishi et al. reported the use of benzamide derivatives with palladium catalysis to achieve ortho-selective chlorination reactions using hydrochloric acid under anodic oxidation conditions
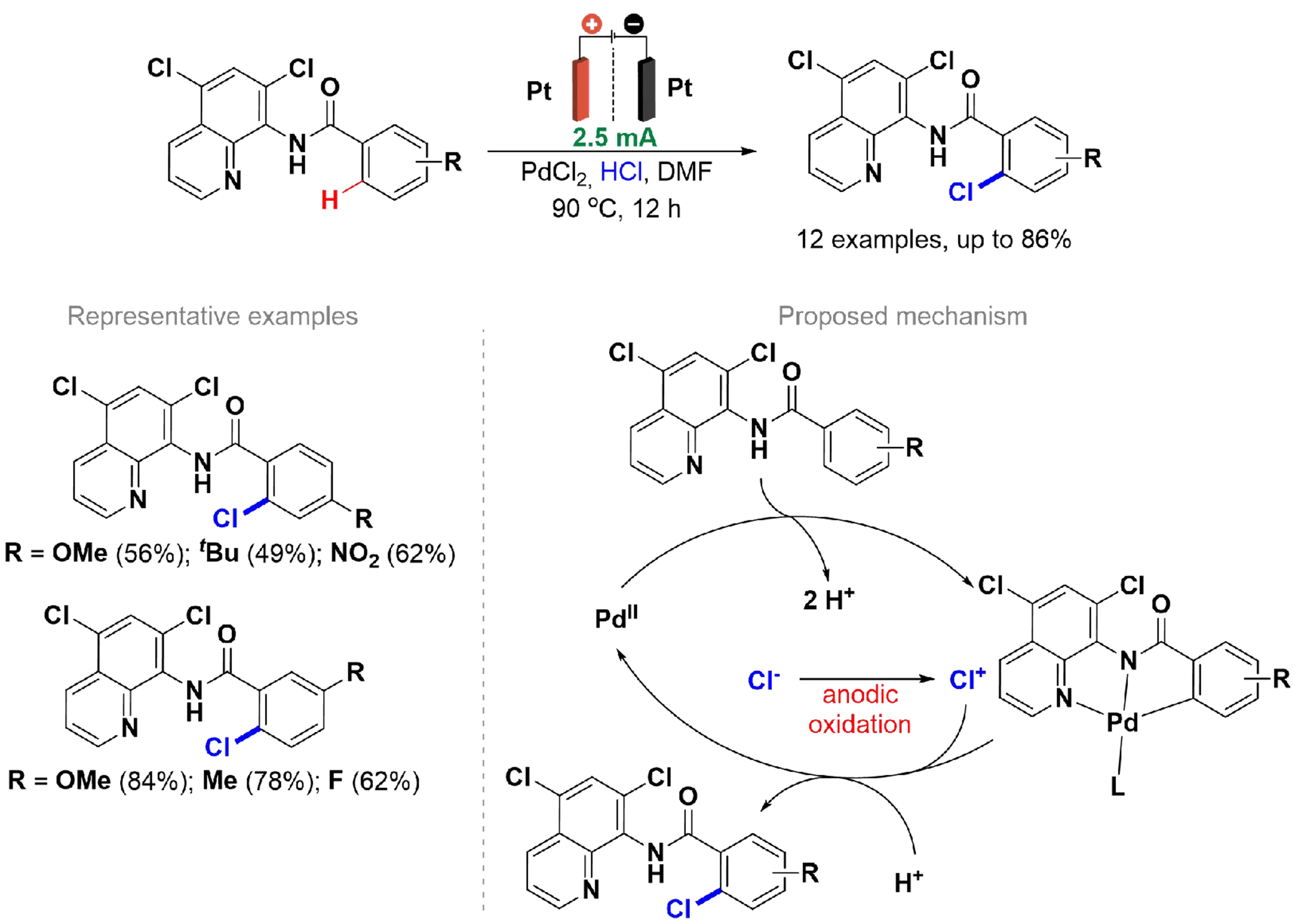
Scheme 16. Pd-catalyzed C(sp2)–H chlorination of benzamide derivatives.
Ashikari et al. reported an electrolysis-based oxidation of diarylmethanes and substituted alkenes containing aryl groups to their corresponding carbonyl compounds using a divided cell equipped with a CF electrode at the anode and a platinum electrode at the cathode [Scheme 17][105]. An alternative method was developed for oxidizing certain substrates to carbonyl compounds, addressing the limitation posed by the lower or equal oxidation potential of DMSO (1.76 V vs. SCE) compared to some substrates. This method follows a cation formation at very low temperatures (-78 °C), with DMSO added in a separate step. Cyclic voltammetry was conducted, and the reaction demonstrated yields of up to 91% on several substrates. Mechanistic studies, including tests with deuterated DMSO, indicated that the substrate is first oxidized at the anode to form a carbocation, which is then attacked by DMSO to yield a more stable alkoxysulfonium ion. The final carbonyl compound is produced by treatment with a base (NR3). The authors referenced findings by Thanh et al., who observed that diarylcarbenium ions generated from SN1 reactions reacted with DMSO, and then with triethylamine treatment producing diaryl ketones[106].
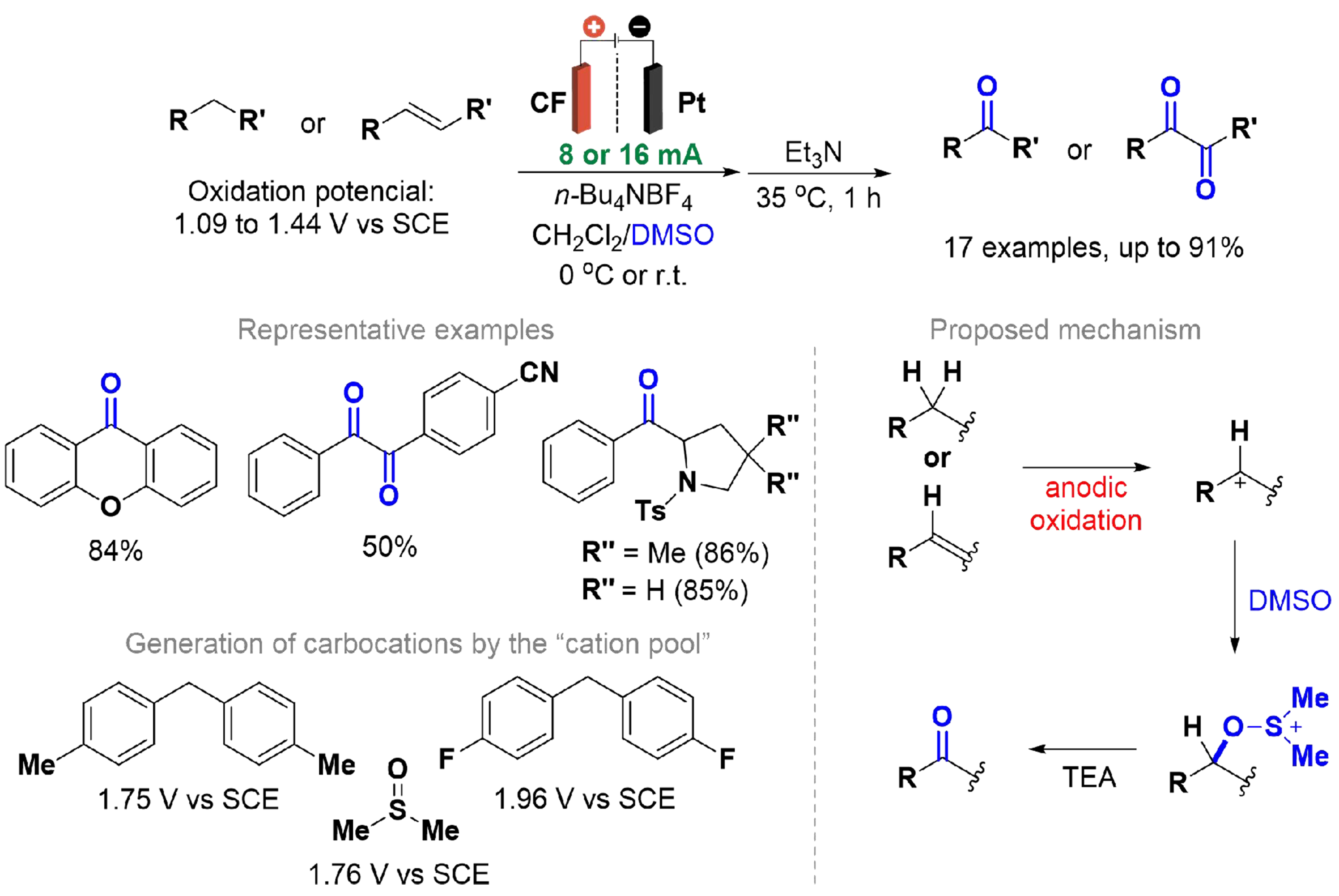
Scheme 17. Oxidation of diarylmethanes and substituted alkenes derivatives.
The production of furfural is well-established, but recent concepts in circular economy have driven research towards improving production methods and diversifying applications[107]. Furfural is recognized as one of the top 30 biomass-derived platform molecules, presenting a promising alternative to fossil fuel-derived materials and contributing to the reduction of greenhouse gas emissions[108]. In 2021, Cao et al. reported the paired electrochemical conversion of furfural to produce 2(5H)-furanone via anodic oxidation to generate furfuryl alcohol and/or hydrofuroin via cathodic reduction [Scheme 18][109]. A continuous flow divided cell with graphite as the anode, lead as the cathode, and an anion exchange membrane was utilized. The electrolysis was conducted in an aqueous mixture of furfural and NaBr, which served as a mediator for anodic oxidation. An anion exchange membrane was essential for reaction efficiency, with its absence significantly decreasing yields. Alternative electrode materials such as nickel and stainless steel were less effective in affording the desired compounds. In the anodic chamber, furfural is oxidized to 2(5H)-furanone through a hydroxyl radical-induced C–C bond cleavage, leading to the formation of 2-hydroxyfuran and formic acid[110]. The 2-hydroxyfuran subsequently isomerizes to 2(5H)-furanone. In the cathodic chamber, furfural is reduced to generate radicals[111], which can dimerize to form hydrofuroin or be further reduced to produce furfuryl alcohol.
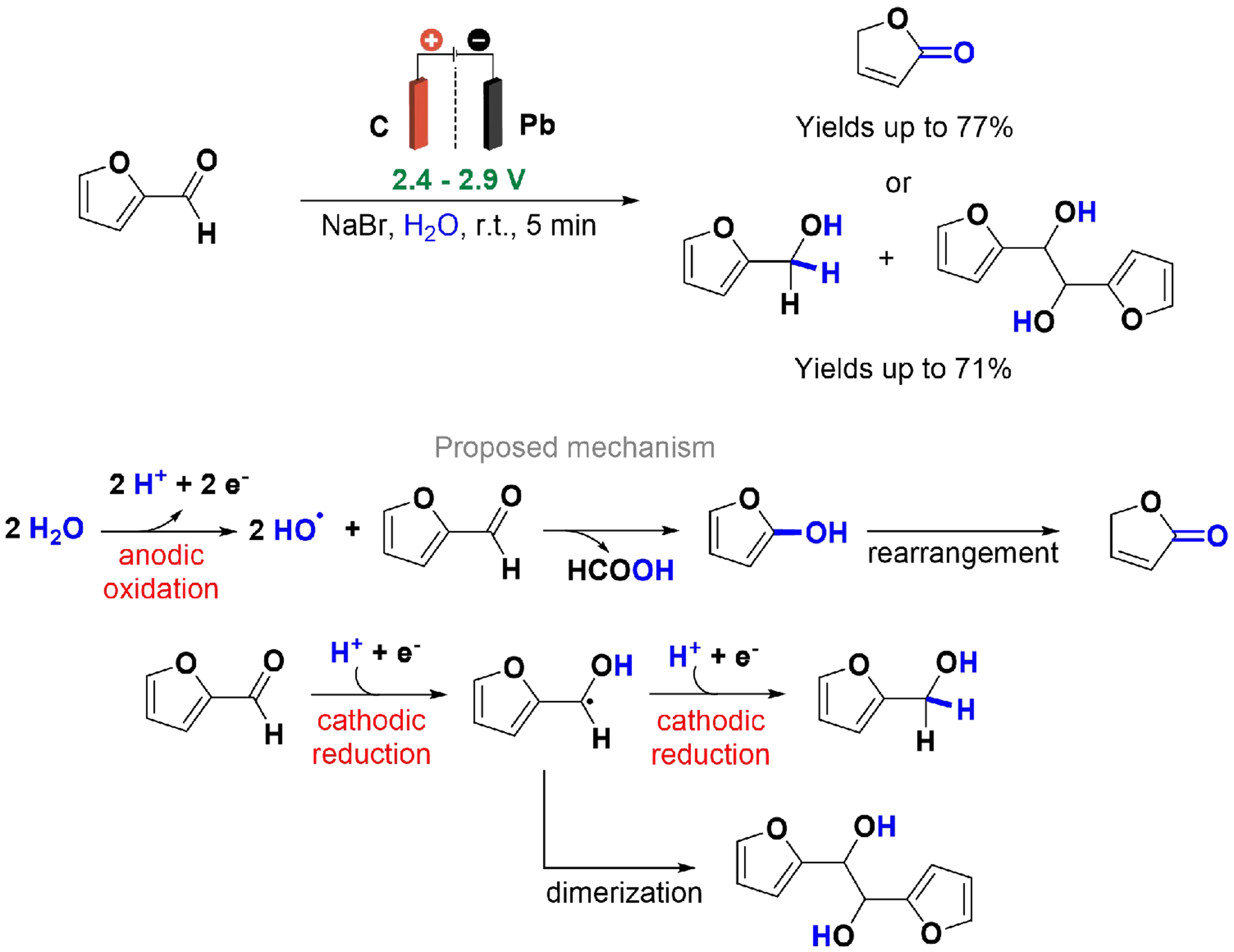
Scheme 18. Paired electrolysis of furfural.
Wu et al. presented a selective semi-hydrogenation and deuteration of several terminal and internal alkynes using H2O and D2O as hydrogen and deuterium sources, employing a Pd-P as cathode electrode
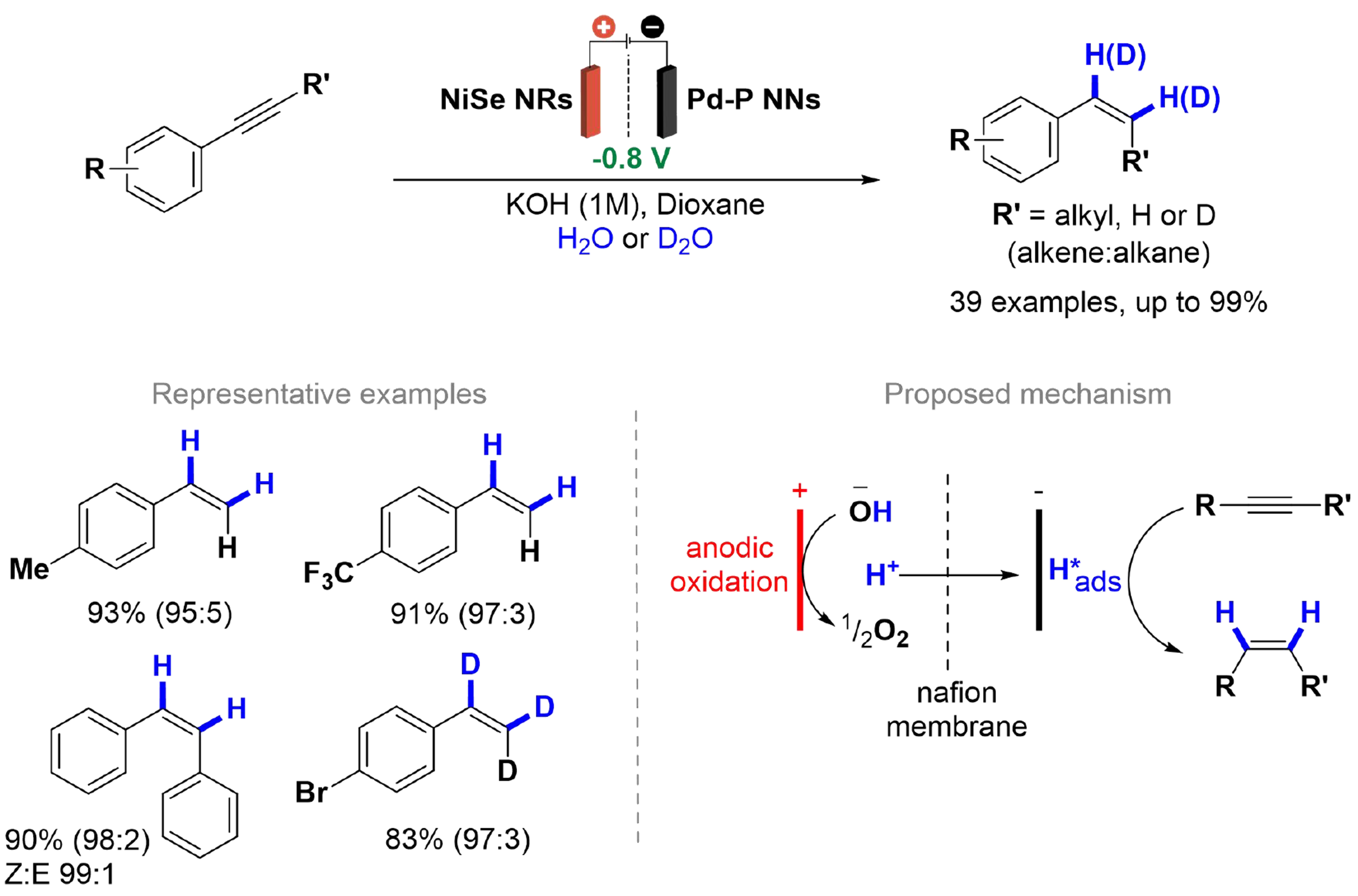
Scheme 19. Semihydrogenation of alkynes with H2O and D2O.
INDIRECT ELECTROLYSIS
Indirect electrolysis enhances the efficiency and selectivity of organic transformations by using mediators or sacrificial electrodes to facilitate selective oxidation or reduction reactions. This method is particularly advantageous for complex transformations, where precise control over reaction conditions can improve yields and minimize side reactions. By carefully selecting mediators, chemists can direct reaction pathways to achieve desired products with high specificity, establishing indirect electrolysis as an essential tool in organic electrosynthesis.
The formation of C–C bonds is crucial in synthetic organic chemistry, especially for the synthesis of natural products and active pharmaceutical ingredients (APIs). However, many methods described in the literature for forming C–C bonds through the functionalization of aromatic C–H bonds rely on reactive groups and metal catalysis[113]. On the other hand, Saito et al. described a Pd-catalyzed homo-coupling method for arylpyridines under anodic oxidation conditions. This method employed an H-type divided cell with an anion-exchange membrane and two platinum electrodes, with reactions performed at a continuous current at 90 °C [Scheme 20][114]. During the optimization of reaction conditions, selective dimerization occurred at the less sterically hindered ortho position, unlike Pd-catalyzed dimerization with oxone[115]. Increasing substrate concentration improved yields while allowing the use of catalytic amounts of I2 as a redox mediator. Without electrical current, the reaction yielded only 31% after five hours, compared to 85% with a current of 20 mA. The proposed mechanism involves the cleavage of ortho C–H bonds, forming a doubly chelated Pd(II) species, which is oxidized by I+ generated from the anodic oxidation of I2.
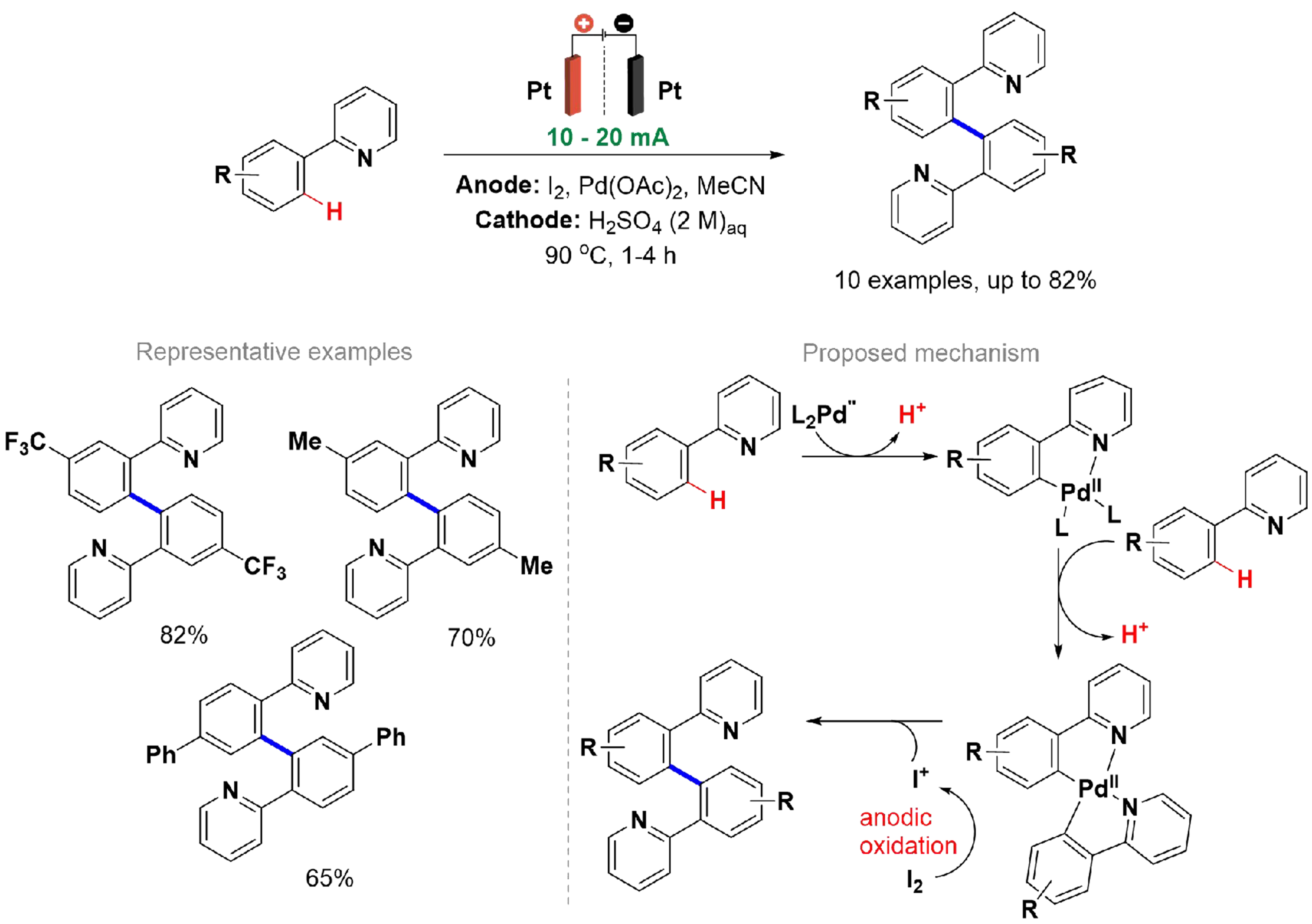
Scheme 20. Palladium-catalyzed homo-coupling of arylpyridines under anodic oxidation conditions.
Palladium-catalyzed C–H cross-coupling reactions have become an important tool for constructing new
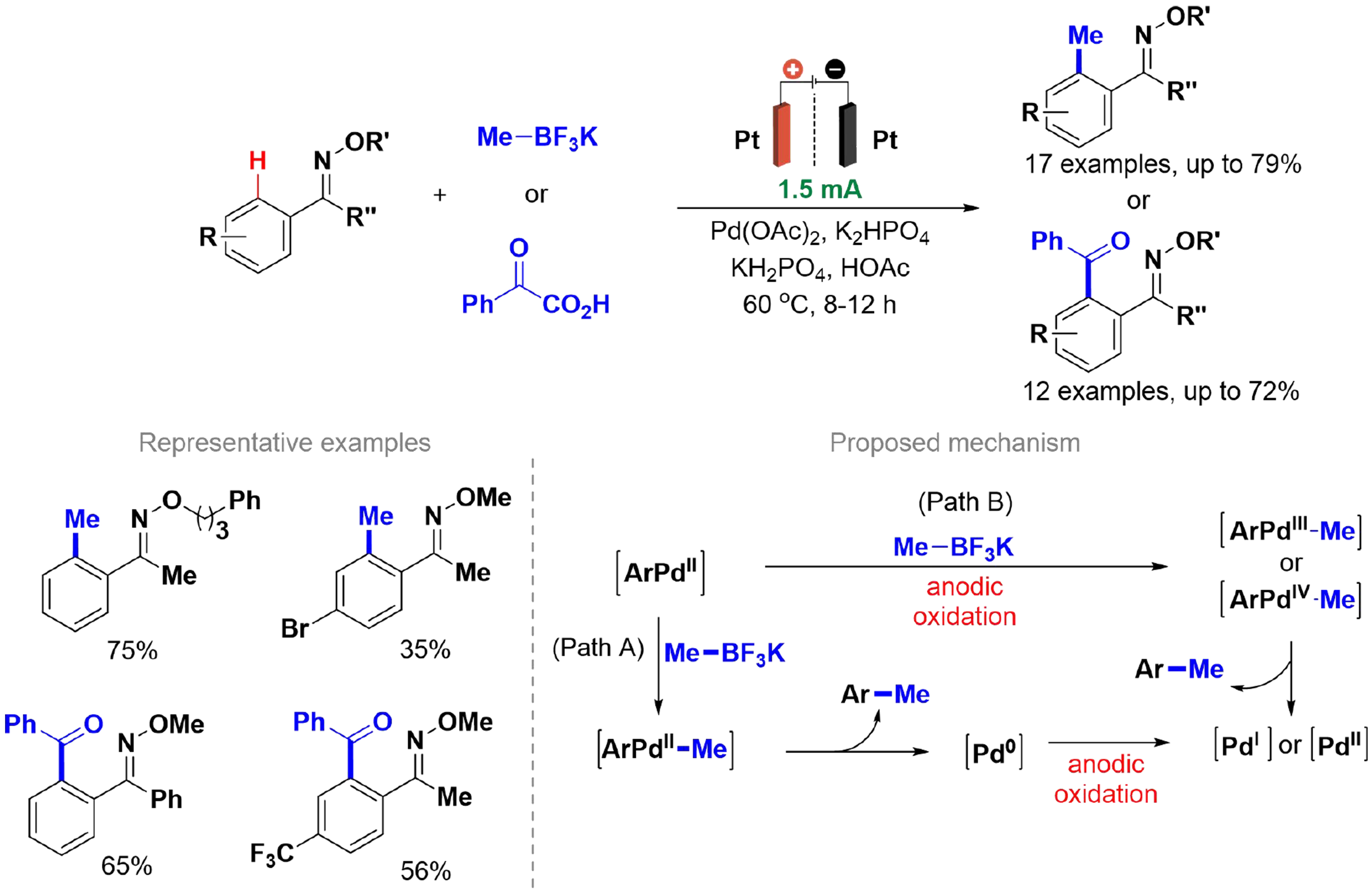
Scheme 21. Pd-catalyzed C(sp2)–H coupling of ketoxime ethers with organoboron compounds or α-ketoacids.
Parameter variation tests were performed, yielding two scopes for methylation and acylation reactions. Methylation results indicated that Cl and Br led to lower yields, suggesting limited tolerance for these substituents. To understand the reaction mechanism, cyclic voltammetry, kinetic tests, and X-ray analysis of the in situ catalyst were conducted. Two proposed mechanisms for C(sp2)–H methylation via electrochemical oxidation involve the palladium catalyst coordinating with the substrate nitrogen to form a palladacycle, which can react through transmetallation to yield Pd(II) or form Pd(III) or Pd(IV) intermediates. Reductive elimination then generates the product and Pd(0), which can be oxidized to regenerate high-valence species. This method shows advantages over other methods since it replaces stoichiometric Cu or Ag salts by electric current to oxidize Pd(0) to Pd(II)[118].
In addition to constructing carbon-carbon bonds, the use of palladium to catalyze C–H activation has emerged as an interesting method to perform carbon-heteroatom bond formation[119]. In 2017, Li et al. reported a Pd-catalyzed C(sp2)–H acetoxylation of ketoxime ethers using electric current instead of a stoichiometric amount of oxidants, following the same principles as before [Scheme 22][120]. A divided cell with two platinum electrodes and an anion exchange membrane was used at 40 °C.
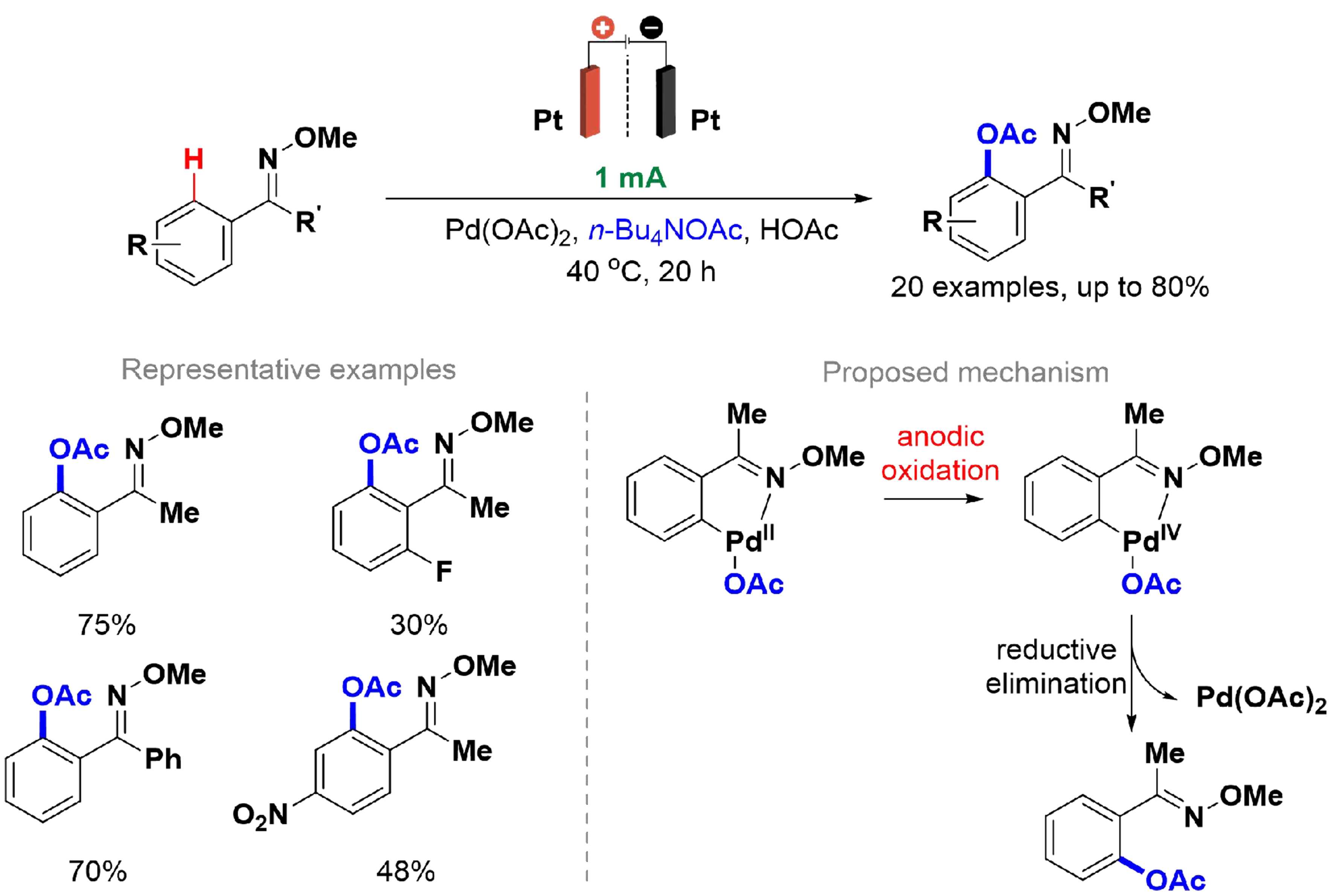
Scheme 22. Pd-catalyzed C(sp2)–H acetoxylation of ketoximes ethers.
Several experiments were conducted to obtain the best reaction conditions, and a relatively large scope was explored. The authors achieved yields of up to 80%, but a low result (30%) was obtained when the substrate was substituted in the ortho-position, due to low conversion. Kinetic evaluations were performed, and a mechanism for C(sp2)–H acetoxylation was proposed. The nitrogen of the ketoxime coordinates with Pd(II) followed by C–H activation (the rate-determining step) to generate the palladacycle. Next, the Pd(II) complex is directly oxidized at the anode to form the Pd(IV) complex and undergoes reductive elimination, giving the desired product and closing the catalytic cycle. In the first report of Pd-catalyzed C(sp2)–H acetoxylation using pyridines, Dick et al. applied stoichiometric amounts of oxidants to promote the oxidation of the Pd(II) complex and a high temperature of 100 °C[121]. Using electric current in a divided cell presents an interesting advantage.
Li et al. developed a method for Friedel-Crafts alkylation of electron-rich aromatics with N-vinylamides using an electrochemically generated tris(p-bromophenyl)amine (TBPA) radical cation, achieving yields of up to 93% [Scheme 23][122]. During electrolysis, TBPA is oxidized and functions as an ET catalyst, initiating a cationic chain reaction. A polymeric ionic liquid-carbon black (PIL-CB) composite was employed as the supporting electrolyte, which is both recoverable and reusable, retaining its efficiency and stability over four electrolysis cycles. After TBPA electrolysis, the electrodes were removed, and the substrates were quickly added to the anode compartment, where they were stirred until the starting materials were completely consumed. Cyclic voltammetry and control experiments suggested that the reaction mechanism involves oxidative activation of the C–H bond in electron-rich aromatics.
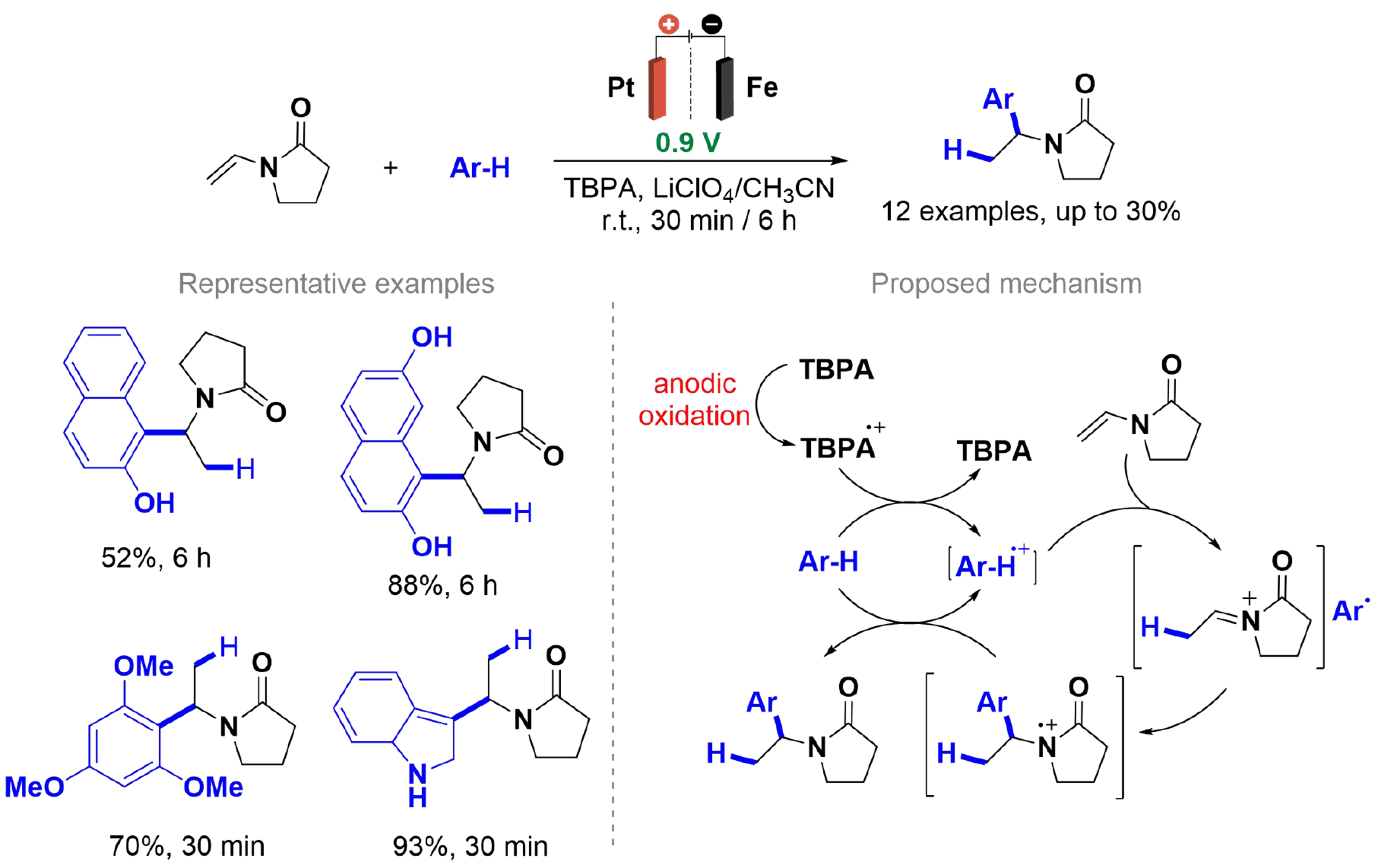
Scheme 23. Friedel-Crafts alkylation of aromatics with vinylamides.
The conversion of aliphatic C–H bonds to carbon-oxygen (C–O) bonds using selective catalytic methods remains a significant challenge in organic synthesis[123]. In 2017, Yang et al. reported a Pd-catalyzed C(sp3)–H oxygenation using different oxyanions of ketoxime ethers under electrosynthetic conditions
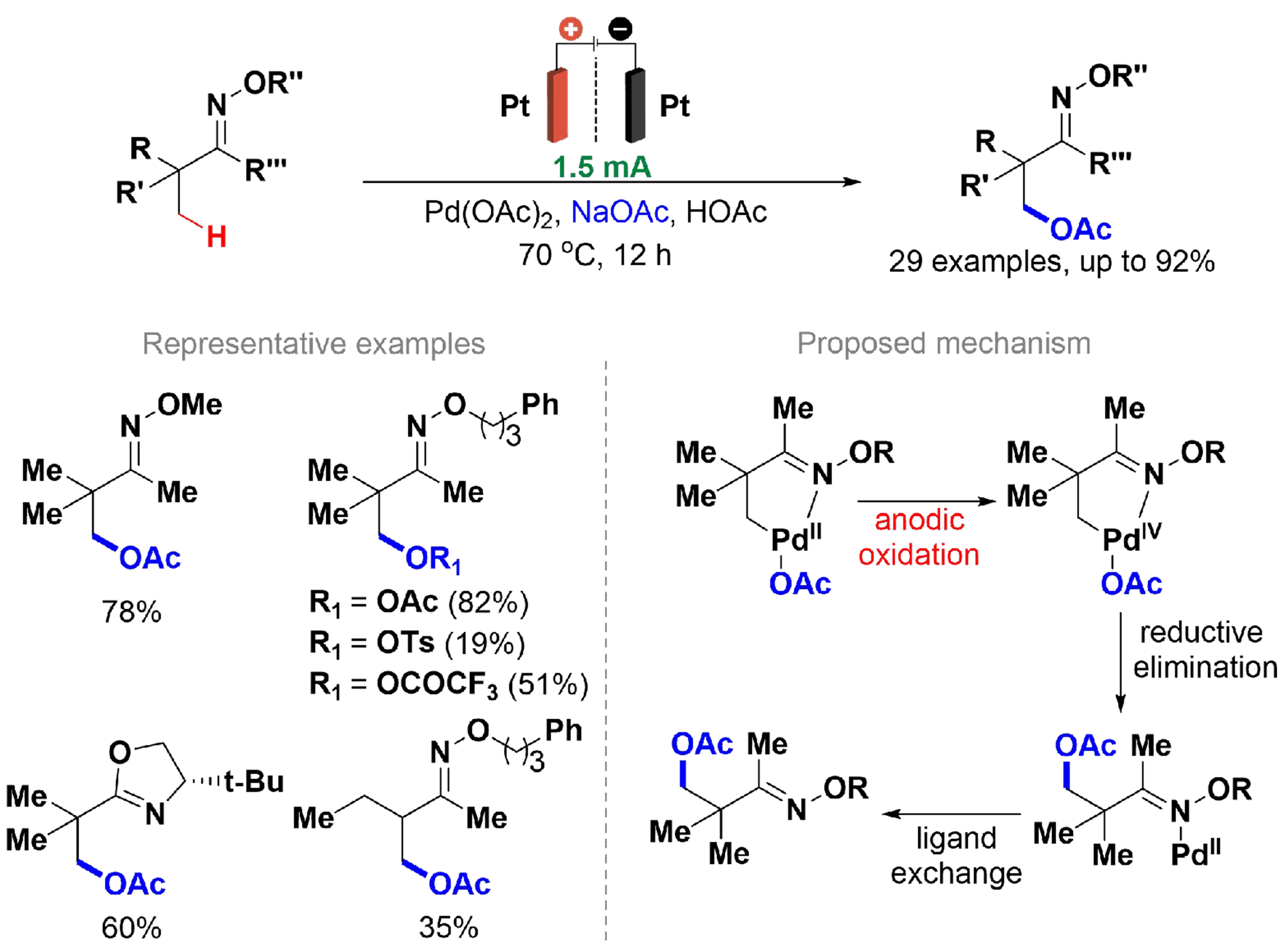
Scheme 24. Pd-catalyzed C(sp3)–H oxygenation of ketoxime ethers.
Robinson et al. developed an electrochemical method for Ru-catalyzed C–H hydroxylation of amine derivatives at tertiary C–H bonds [Scheme 25][125]. By applying electric current, the need for a stoichiometric chemical oxidant was eliminated without compromising catalyst efficiency. The cis-[Ru(dtbpy)2Cl2] catalyst demonstrated stability in aqueous acidic media, and the use of a divided cell provided proton reduction as a cathodic reaction without interference and eliminated the need for supporting electrolytes. The results were comparable to those previously reported by the same authors, which relied on stoichiometric H5IO6 as the oxidant[126]. Despite their limited solubility, various functional groups such as amides, imides, benzoyl-protected alcohols, and electron-deficient arenes have been effectively oxidized electrochemically. Mechanistic studies involving a Ru complex revealed redox processes between Ru(IV)/(V) and Ru(V)/(VI), indicating that catalytically active species such as oxo- or dioxo-Ru(V) or Ru(VI) intermediates can be generated through electrochemical oxidation. Cyclic voltammetry of the Ru(II) catalyst showed that five distinct oxidation states [Ru(II)–Ru(VI)] are accessible electrochemically within 800 mV in acidic aqueous solutions.
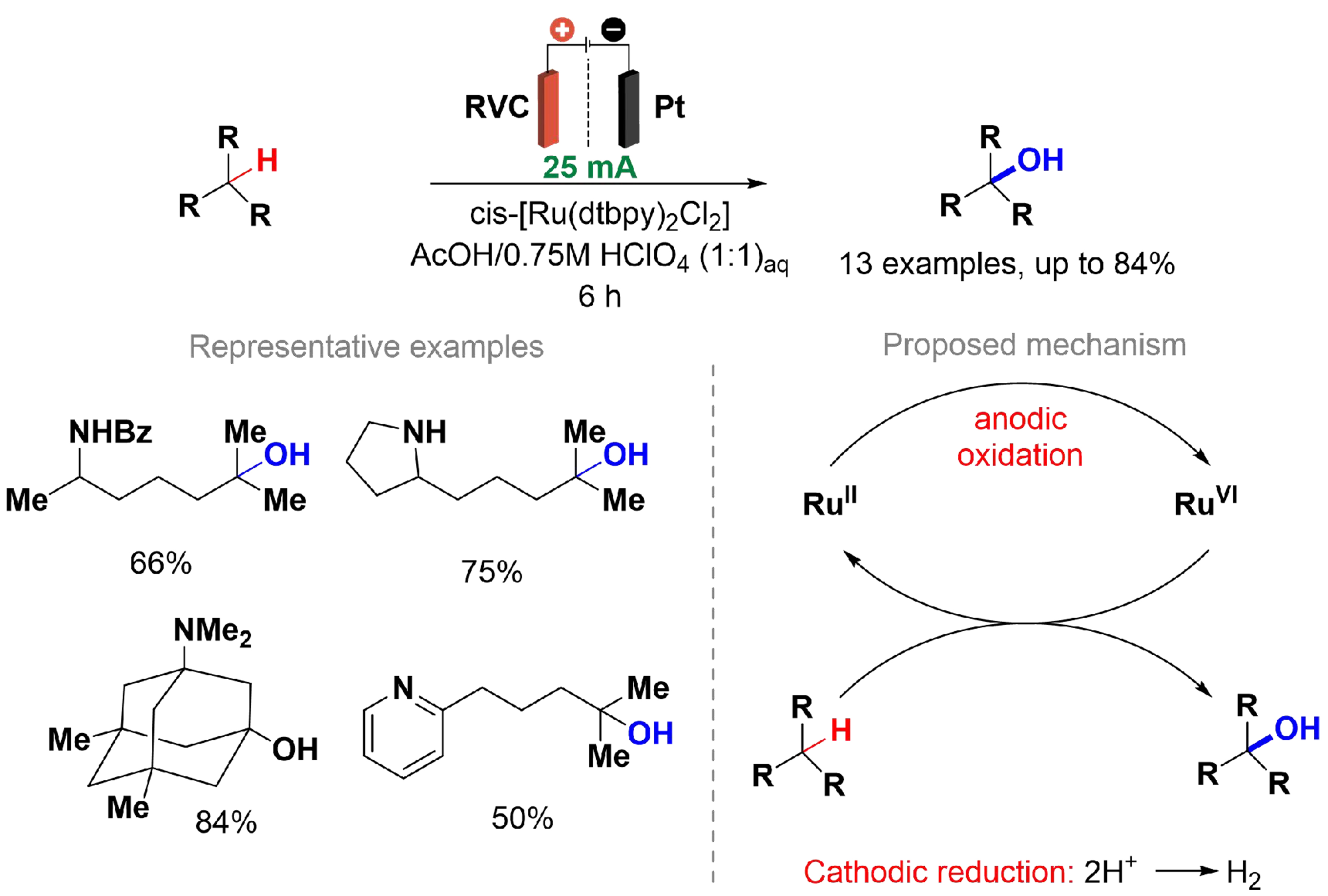
Scheme 25. Ruthenium-catalyzed C–H hydroxylation of amine derivatives.
Ethylene oxide is one of the most highly produced chemicals globally, widely utilized in the manufacture of plastics, detergents, solvents, and other products, with annual production surpassing millions of tons[127]. In 2020, Leow et al. developed an electrochemical methodology for the synthesis of epoxides from the corresponding olefins [Scheme 26][17]. A continuous flow divided cell was applied, featuring a platinum anode, nickel cathode, and an anion exchange membrane. Using a 1.0 M potassium chloride (KCl) solution as the electrolyte and varying current densities from 0.3 to 1.0 A/cm2, yields reached up to 71%. This method avoids CO2 emissions, as the chloride electrolyte mediates the reaction by converting to chlorine gas (Cl2), forming hypochlorous acid (HOCl) and hydrochloric acid (HCl). Ethylene reacts with HOCl to produce chlorohydrin, while water reduction at the cathode generates H2 and hydroxide anions, which further react with chlorohydrin and HCl to yield the desired product.
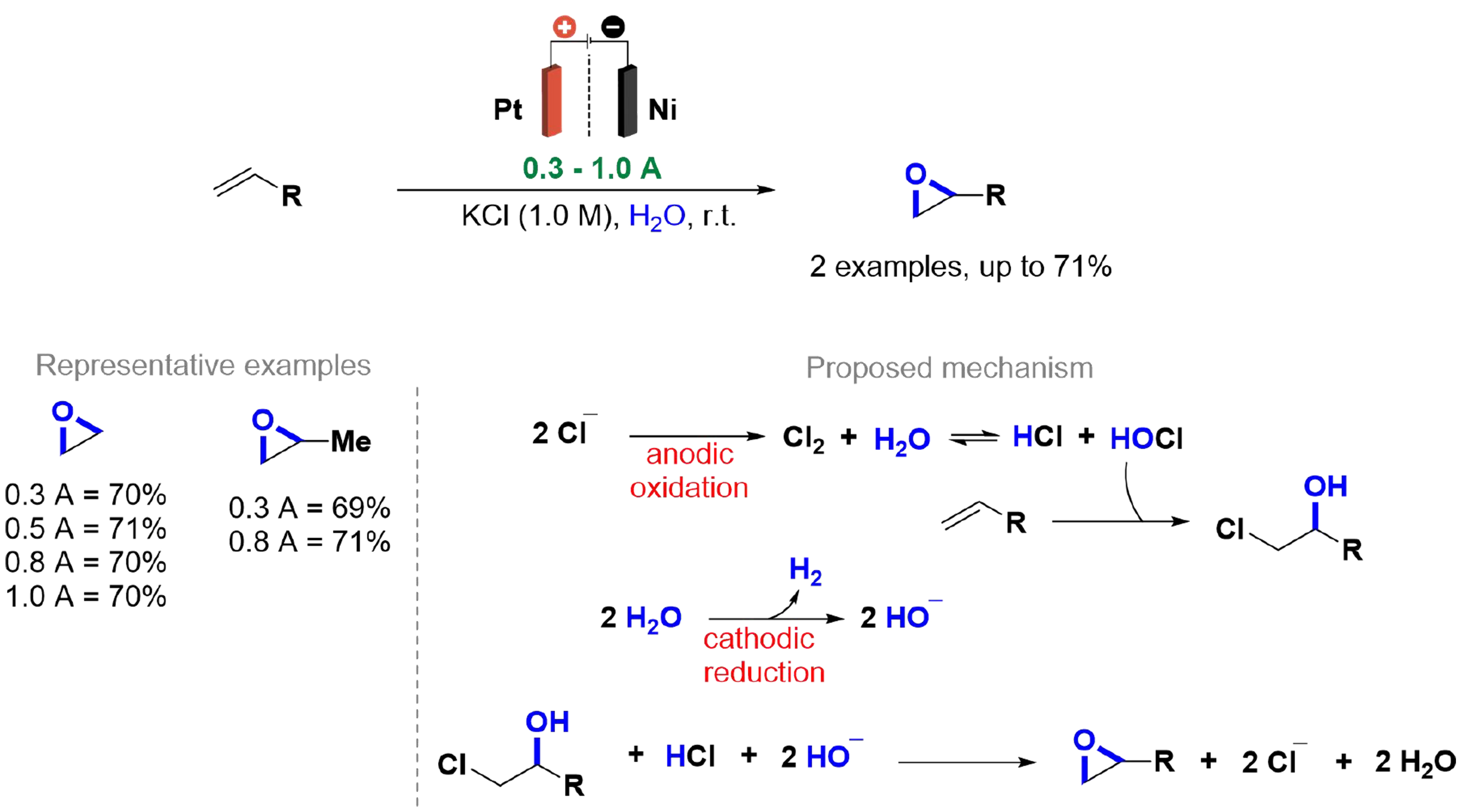
Scheme 26. Chloride-mediated selective electrosynthesis of epoxides.
Rafiee et al. reported a benzylic C–H iodination of several methylarenes, driven by the electrochemical oxidation of N-hydroxyphthalimide (NHPI) to phthalimido-N-oxyl (PINO), which mediates hydrogen atom transfer (HAT) to generate benzylic radicals [Scheme 27][128]. The reaction was conducted in an H-type divided cell with a vitreous carbon (VC) anode and platinum wire cathode. Imidoxyl mediators, such as
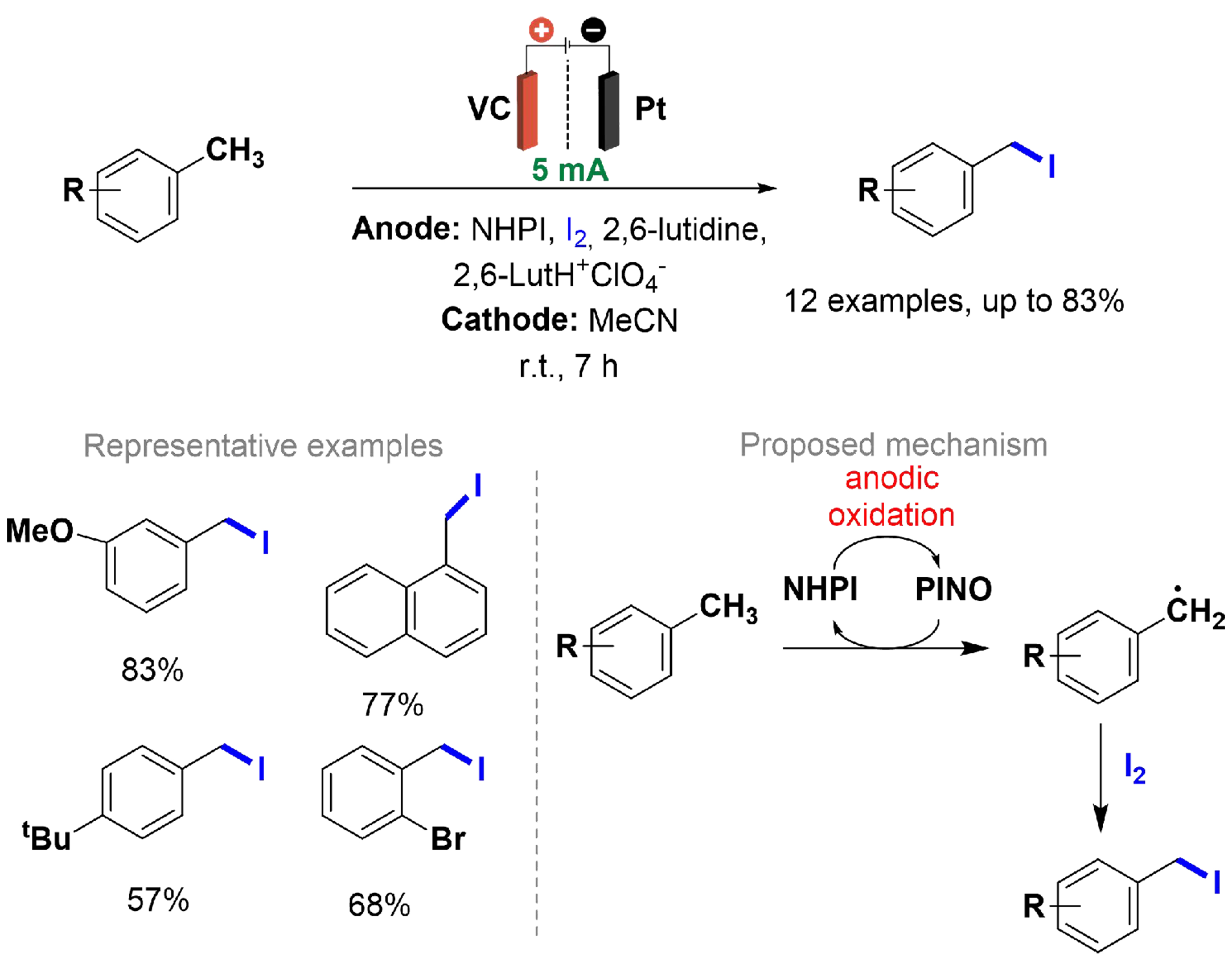
Scheme 27. C(sp2)–H iodination of methylarenes.
The reductive cleavage to generate aryl radicals can be achieved through direct electrolysis at high potentials or by employing alkali metals[129,130]. However, both methods have limitations regarding yield and chemoselectivity. Electrochemical stimulation via single ET (SET) processes in organic molecules offers electron-primed photoredox catalysts, which can efficiently and safely cleave strong bonds, such as C(sp2)–N and C(sp2)–O, with high potentials while maintaining chemoselectivity. In this context, Chernowsky et al. reported a defunctionalization reaction utilizing 2,4,5,6-Tetrakis(diphenylamino)isophthalonitrile (4-DPAIPN) as an electron-primed photoredox catalyst in electrolysis using a divided cell with a glass frit, employing an RVC cathode and anode [Scheme 28][131]. A broad substrate scope was evaluated, revealing that several functional groups are well-tolerated, allowing for the cleavage of C(sp2)–N bonds from anilinium salts and even higher energy C(sp2)–O bonds from phosphate ester substrates and phenols. Studies on cathodic potentials and cyclic voltammetry demonstrate that the reaction proceeds only when the applied potential is sufficient to reduce the photoredox catalyst 4-DPAIPN, indicating that cathodic reduction catalysis is essential for the reaction. In this process, the catalyst is reduced at the cathode, forming a radical anion species that is subsequently excited by visible light. This excitation leads to a highly reduced state, termed “electron-primed”, which is capable of cleaving C(sp2)–N and C(sp2)–O bonds, generating aryl radicals that can be effectively trapped. The cathodic reduction is highly reversible, and the use of a divided cell electrolysis setup prevents interference from oxidized byproducts.
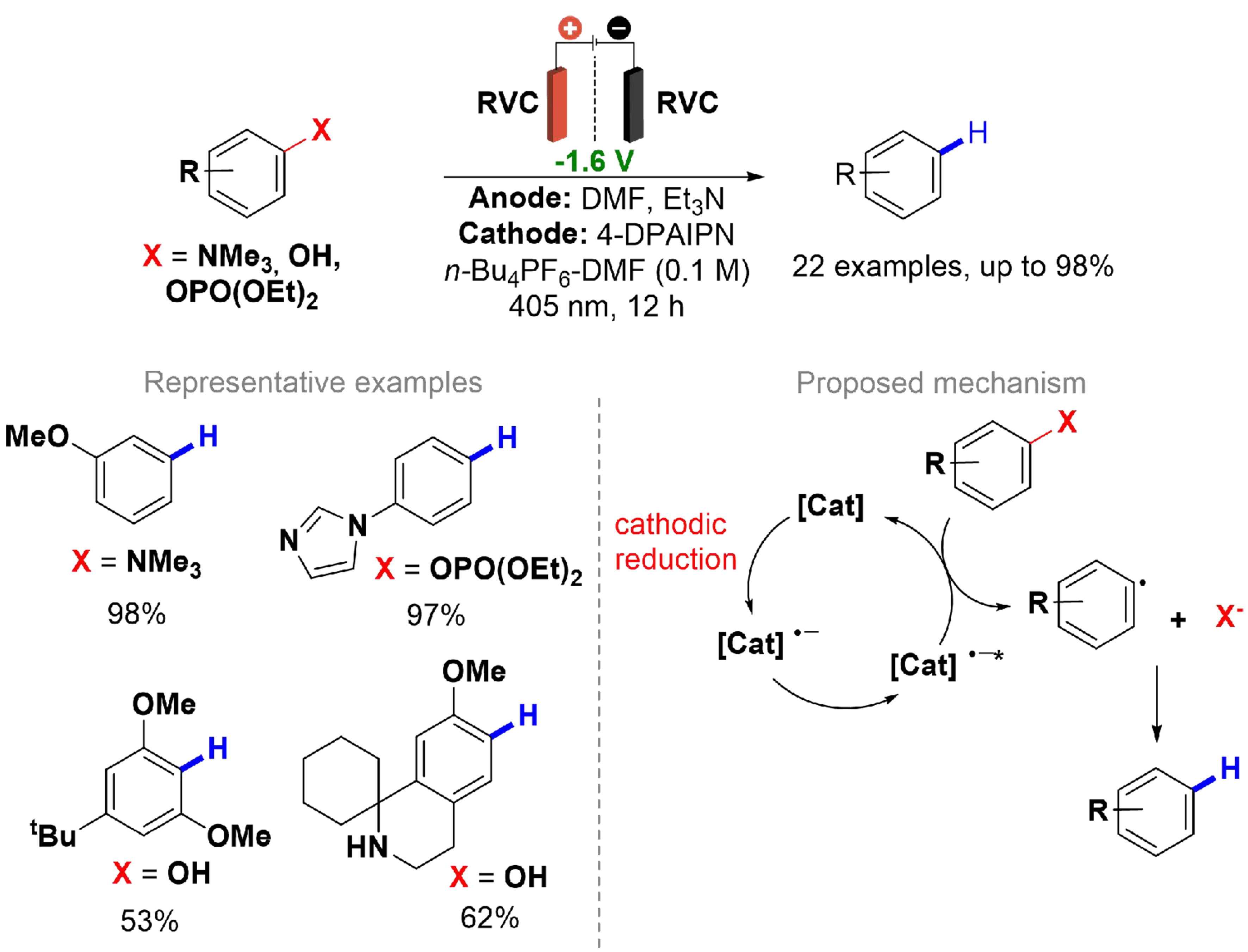
Scheme 28. Electrochemical generation of electron-primed photoredox catalyst for cleavage of C(sp2)–N and C(sp2)–O bonds.
In 2020, Siewert and Fokin introduced an electrochemical method for the chemoselective reduction of ketones and aldehydes using a manganese catalyst [Scheme 29][132]. This method uses protons and electric current to convert carbonyl groups into alcohols with high efficiency and selectivity. The setup involved carbon foam as the working electrode, a platinum wire as the counter electrode, and an Ag/AgNO3 reference electrode. All experiments were carried out in a glovebox within a divided cell. Manganese centers enhance proton transfer during the reduction, improving reaction performance. Under optimized conditions, the process delivers high conversion rates and selectivity. Hydride species (Mn-H) are generated in situ via the electrochemical reduction and protonation of the manganese complex, enabling selective reduction of ketones and aldehydes.
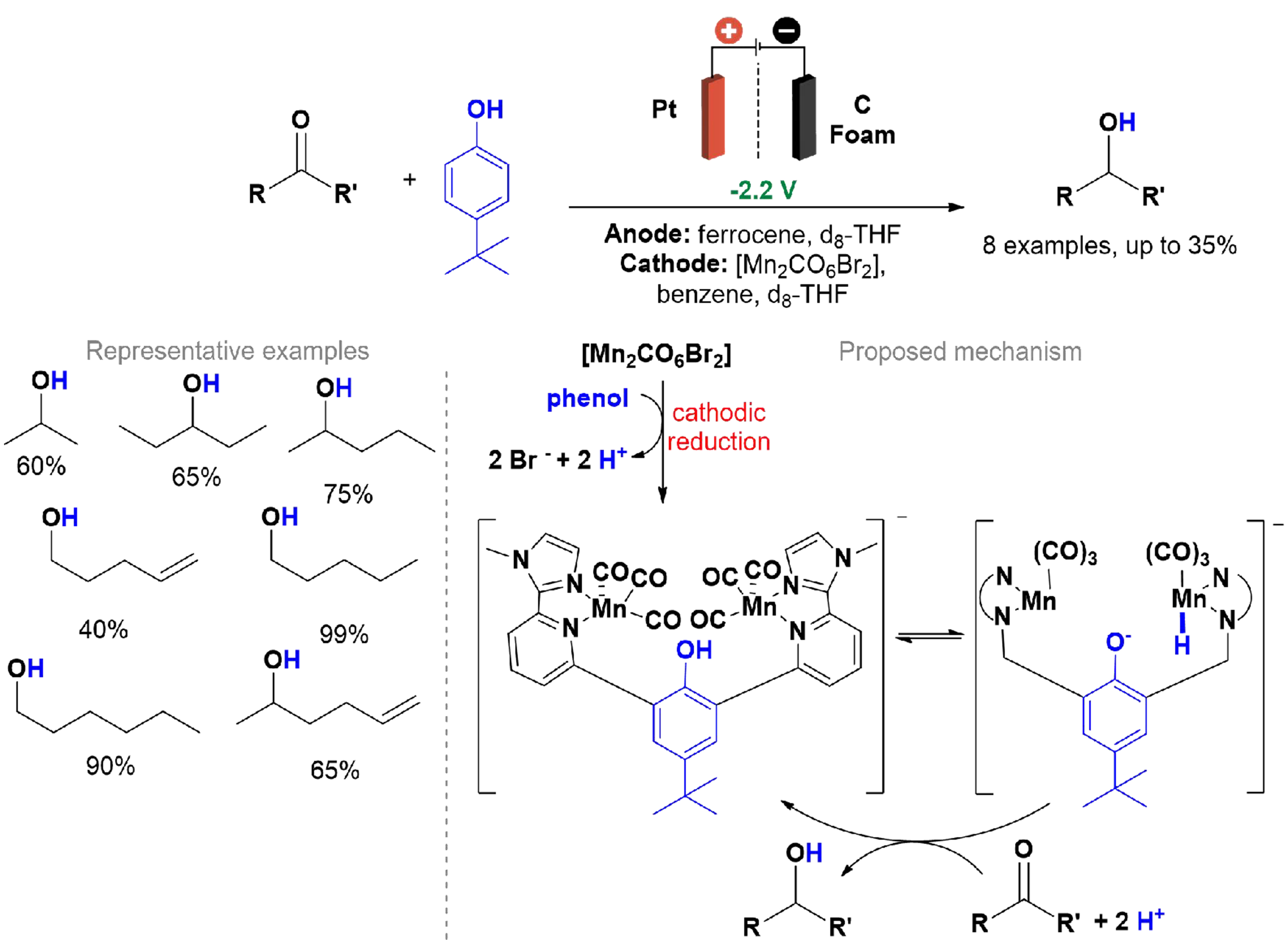
Scheme 29. Reduction of ketones and aldehydes using electrochemically activated Mn catalyst.
Hoque et al. present an innovative approach to the activation of molecular oxygen (O2) through coupled electrochemical reactions that combine O2 reduction at the cathode and water oxidation at the anode, utilizing a manganese-porphyrin catalyst and VC electrodes [Scheme 30][133]. This process facilitates the formation of reactive manganese-oxo species at both electrodes, enabling oxygen atom transfer (OAT) to organic compounds, such as sulfides, which are converted selectively into sulfoxides with high Faradaic efficiency.
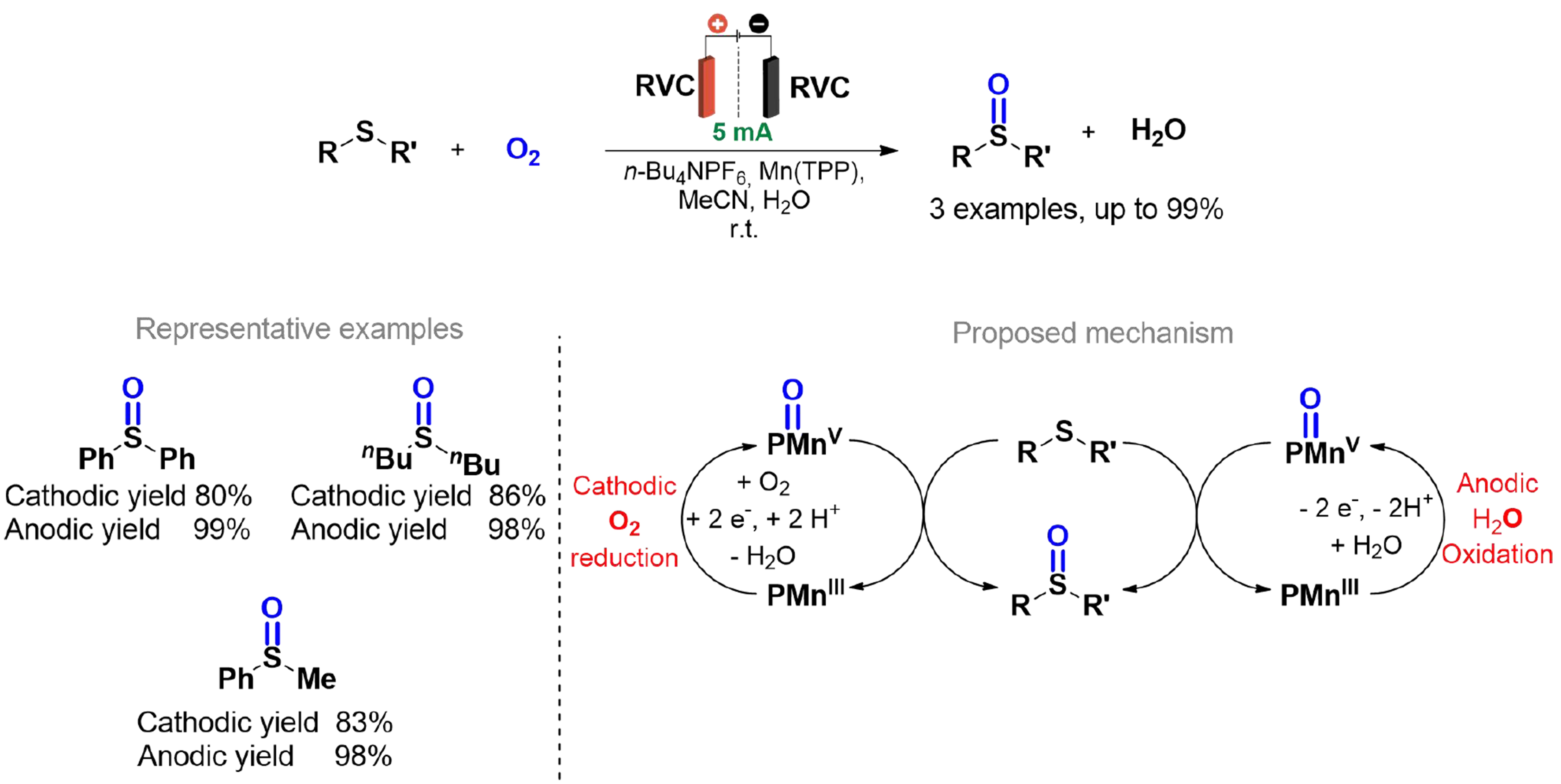
Scheme 30. Oxidation of sulfides using electrochemically activated Mn catalyst.
The use of an H-type divided cell was crucial to optimize the process, ensuring effective separation of anodic and cathodic reactions, preventing competitive side reactions, and increasing Faradaic efficiency up to 200%. This methodology represents a sustainable and efficient strategy to overcome kinetic barriers in OAT reactions without net electron consumption, demonstrating applicability to a wide range of organic oxidation reactions, including epoxidations and C–H hydroxylations. These advancements underscore the potential of electrochemical energy in organic synthesis and energy conversion technologies.
ENZYMATIC AND MICROBIAL ELECTROCHEMICAL PROCESSES
In recent decades, significant efforts have been dedicated to studying enzymatic systems for applications in biosensors, energy conversion, biofuel cells, and biocapacitors[134]. In enzymatic biocatalysis, DET between redox enzymes and electrodes[135], enabled by functionalized surfaces with conductive materials such as metallic nanoparticles and redox polymers, is a critical mechanism [Figure 4][37]. The electrochemical regeneration of redox cofactors, such as NAD(P)H, and strategies based on mediated ET (MET) using redox mediators enhance efficiency and overcome kinetic limitations[136].
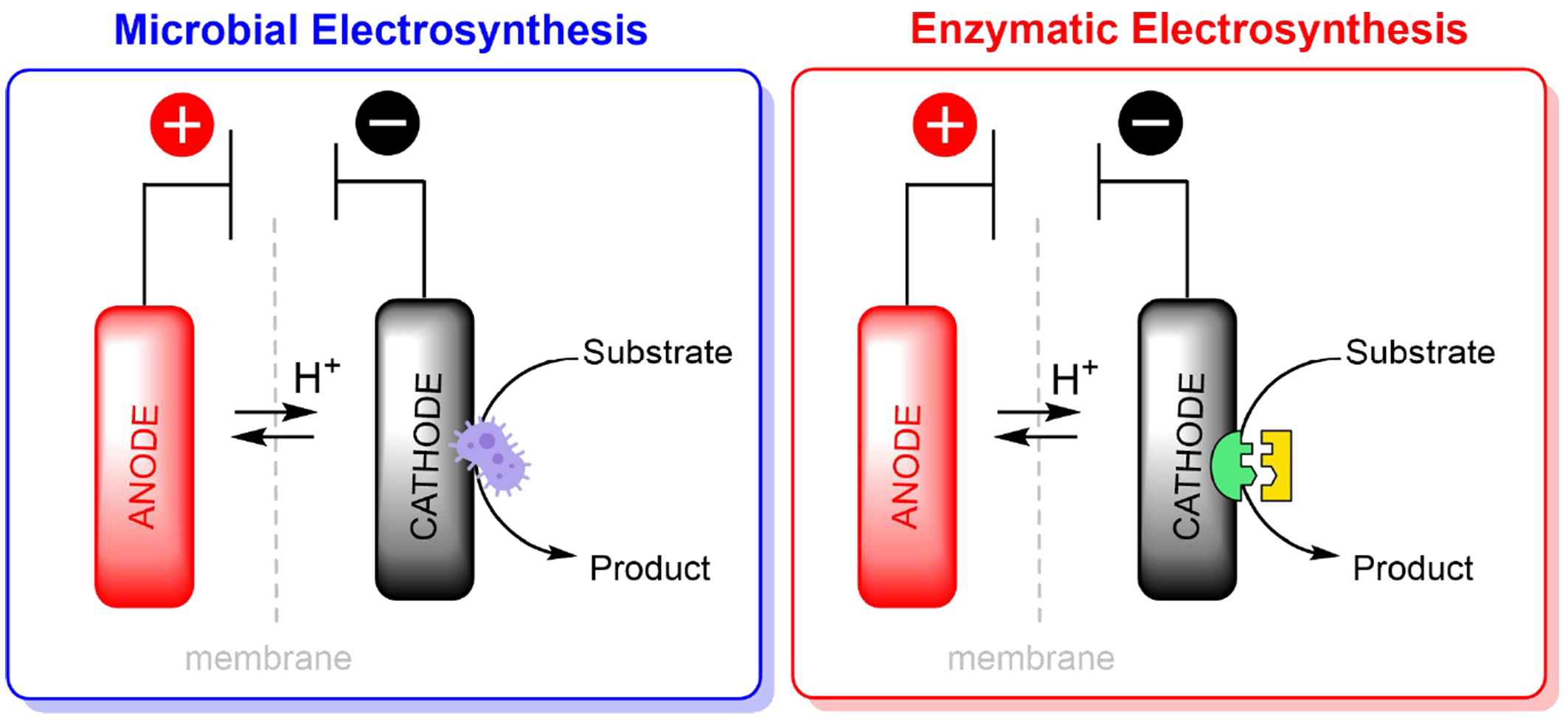
Figure 4. General bioelectrocatalytic synthetic systems.
In microbial systems, extracellular ET (EET) is mediated by structures such as cytochromes and conductive nanowires, which connect intracellular metabolic machinery to electrodes[137]. Electroactive microorganisms, such as Geobacter sulfurreducens and Shewanella oneidensis, have been widely explored for the bioelectrochemical conversion of CO2 and organic compounds into high-value products, including carboxylic acids and biofuels[138].
Although integrating biocatalysts with electrochemical systems poses challenges[139], such as mass transport limitations, side reactions, and biocatalyst stability, advances in high-surface-area electrodes and divided cells have significantly improved the efficiency and feasibility of these processes[140-143].
CONCLUSIONS AND OUTLOOK
Electrosynthesis reactions have demonstrated substantial advantages for several organic transformations. By using electricity as a clean reagent, divided cells enable controlled redox transformations with high selectivity, minimizing undesired byproducts and providing distinct benefits for complex organic reactions, such as selective C–H functionalization, oxidative dehydrogenation, and cross-coupling. These methodologies address challenges often encountered with traditional methods, including cross-contamination and side reactions.
Divided cell setups offer unique capabilities, allowing independent optimization of anodic and cathodic reactions. This design enhances both reaction specificity and product isolation while confining reactive intermediates to their respective compartments, preventing undesired recombination and improving overall efficiency. However, the implementation of divided cells also introduces limitations, such as higher costs and complexities associated with membrane use, particularly Nafion membranes, which are crucial for their selectivity and stability. Overcoming these economic and technical barriers through the development of cost-effective and durable alternatives will be essential for wider adoption. Additionally, challenges such as increased internal resistance and difficulties in industrial scaling must be addressed to optimize the energy demands and operational feasibility of these systems.
Looking ahead, innovations in electrode materials, particularly those that are sustainable and robust, will be critical for enhancing the performance and reliability of divided cell electrosynthesis. Advances in membrane technology, including fouling prevention, improved ion selectivity, and cost reductions, could transform the scalability and affordability of these systems. Furthermore, the development of commercial cells adaptable to various scales, including continuous flow reactors, holds great potential to expand the applicability of this technology to diverse and challenging reactions.
In conclusion, divided cell electrosynthesis embodies a promising approach aligned with the principles of green chemistry, offering versatile applications in organic synthesis. By addressing current limitations and focusing on technological advancements, this method has the potential to become a cornerstone of sustainable chemistry, driving innovation across both academic and industrial domains.
DECLARATIONS
Authors’ contributions
Design and revision of the manuscript: Martins, G. M.; Brocksom, T. J.; de Oliveira, K. T.
Discussion and preparation of the manuscript: Magalhães, M. F. A.; Simoso, G. B.; de Borba, E. S.; Martins, G. M.
Availability of data and materials
Not applicable.
Financial support and sponsorship
The authors would like to thank the São Paulo Research Foundation, FAPESP grants 2013/07276-1 (CEPOF), 2023/04020-8 (de Oliveira, K. T.), 2022/10687-2 (Magalhães, M. F. A.), the Conselho Nacional de Pesquisa - CNPq (de Oliveira, K. T. research fellowship 303333/2022-7), and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) - Financial Code 001.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Yuan, Y.; Lei, A. Is electrosynthesis always green and advantageous compared to traditional methods? Nat. Commun. 2020, 11, 802.
2. Zhu, C.; Ang, N. W. J.; Meyer, T. H.; Qiu, Y.; Ackermann, L. Organic electrochemistry: molecular syntheses with potential. ACS. Cent. Sci. 2021, 7, 415-31.
3. Cabana, J.; Alaan, T.; Crabtree, G. W.; et al. NGenE 2021: electrochemistry is everywhere. ACS. Energy. Lett. 2022, 7, 368-74.
5. Petersen, H. A.; Myren, T. H. T.; O’Sullivan, S. J.; Luca, O. R. Electrochemical methods for materials recycling. Mater. Adv. 2021, 2, 1113-38.
6. Horn, E. J.; Rosen, B. R.; Baran, P. S. Synthetic organic electrochemistry: an enabling and innately sustainable method. ACS. Cent. Sci. 2016, 2, 302-8.
7. Möhle, S.; Zirbes, M.; Rodrigo, E.; Gieshoff, T.; Wiebe, A.; Waldvogel, S. R. Modern electrochemical aspects for the synthesis of value-added organic products. Angew. Chem. Int. Ed. Engl. 2018, 57, 6018-41.
8. Liu, C.; Li, R.; Zhou, W.; et al. Selectivity origin of organic electrosynthesis controlled by electrode materials: a case study on pinacols. ACS. Catal. 2021, 11, 8958-67.
9. Hanssen, B. L.; Siraj, S.; Wong, D. K. Recent strategies to minimise fouling in electrochemical detection systems. Rev. Anal. Chem. 2016, 35, 1-28.
10. Alkhadra, M. A.; Su, X.; Suss, M. E.; et al. Electrochemical methods for water purification, ion separations, and energy conversion. Chem. Rev. 2022, 122, 13547-635.
11. Ahoutou, Y.; Ilinca, A.; Issa, M. Electrochemical cells and storage technologies to increase renewable energy share in cold climate conditions - a critical assessment. Energies 2022, 15, 1579.
12. Krivik, P.; Baca, P. Electrochemical energy storage. In: Zobaa A, editor. Energy storage - technologies and applications. InTech; 2013.
13. Yan, M.; Kawamata, Y.; Baran, P. S. Synthetic organic electrochemistry: calling all engineers. Angew. Chem. Int. Ed. Engl. 2018, 57, 4149-55.
14. Paidar, M.; Fateev, V.; Bouzek, K. Membrane electrolysis - history, current status and perspective. Electrochim. Acta. 2016, 209, 737-56.
15. Lodh, J.; Paul, S.; Sun, H.; Song, L.; Schöfberger, W.; Roy, S. Electrochemical organic reactions: a tutorial review. Front. Chem. 2022, 10, 956502.
16. Hilt, G. Basic strategies and types of applications in organic electrochemistry. ChemElectroChem 2020, 7, 395-405.
17. Leow, W. R.; Lum, Y.; Ozden, A.; et al. Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density. Science 2020, 368, 1228-33.
18. Marken, F.; Cresswell, A. J.; Bull, S. D. Recent advances in paired electrosynthesis. Chem. Rec. 2021, 21, 2585-600.
19. Sherbo, R. S.; Delima, R. S.; Chiykowski, V. A.; Macleod, B. P.; Berlinguette, C. P. Complete electron economy by pairing electrolysis with hydrogenation. Nat. Catal. 2018, 1, 501-7.
20. Sherbo, R. S.; Kurimoto, A.; Brown, C. M.; Berlinguette, C. P. Efficient electrocatalytic hydrogenation with a palladium membrane reactor. J. Am. Chem. Soc. 2019, 141, 7815-21.
21. Llorente, M. J.; Nguyen, B. H.; Kubiak, C. P.; Moeller, K. D. Paired electrolysis in the simultaneous production of synthetic intermediates and substrates. J. Am. Chem. Soc. 2016, 138, 15110-3.
22. Wu, T.; Nguyen, B. H.; Daugherty, M. C.; Moeller, K. D. Paired electrochemical reactions and the on-site generation of a chemical reagent. Angew. Chem. Int. Ed. Engl. 2019, 58, 3562-5.
23. Meyer, T. H.; Choi, I.; Tian, C.; Ackermann, L. Powering the future: how can electrochemistry make a difference in organic synthesis? Chem 2020, 6, 2484-96.
24. Reidell, A. C.; Pazder, K. E.; LeBarron, C. T.; Stewart, S. A.; Hosseini, S. Modified working electrodes for organic electrosynthesis. ACS. Org. Inorg. Au. 2024, 4, 579-603.
25. Gombos, L. G.; Nikl, J.; Waldvogel, S. R. Dual roles of supporting electrolytes in organic electrosynthesis. ChemElectroChem 2024, 11, e202300730.
26. Kishioka, S. Evaluation of formal redox potential from Nernstian plots using higher-order derivative spectra with no background correction. J. Electroanal. Chem. 2023, 930, 117151.
27. Nutting, J. E.; Gerken, J. B.; Stamoulis, A. G.; Bruns, D. L.; Stahl, S. S. “How should I think about voltage? What is overpotential?”: Establishing an organic chemistry intuition for electrochemistry. J. Org. Chem. 2021, 86, 15875-85.
28. Dutta, N.; Bagchi, D.; Chawla, G.; Peter, S. C. A guideline to determine faradaic efficiency in electrochemical CO2 reduction. ACS. Energy. Lett. 2024, 9, 323-8.
29. Medici, F.; Resta, S.; Andolina, S.; Benaglia, M. Recent advances in enantioselective catalytic electrochemical organic transformations. Catalysts 2023, 13, 944.
30. Okada, Y.; Chiba, K. Redox-tag processes: intramolecular electron transfer and its broad relationship to redox reactions in general. Chem. Rev. 2018, 118, 4592-630.
31. Yoshida, J.; Suga, S.; Suzuki, S.; Kinomura, N.; Yamamoto, A.; Fujiwara, K. Direct oxidative carbon−carbon bond formation using the “cation pool” method. 1. Generation of iminium cation pools and their reaction with carbon nucleophiles. J. Am. Chem. Soc. 1999, 121, 9546-9.
32. Shimizu, A.; Takeda, K.; Mishima, S.; et al. Generation, characterization, and reactions of thionium ions based on the indirect cation pool method. Bull. Chem. Soc. Jpn. 2016, 89, 61-6.
33. Suga, S.; Suzuki, S.; Yamamoto, A.; Yoshida, J. Electrooxidative generation and accumulation of alkoxycarbenium ions and their reactions with carbon nucleophiles. J. Am. Chem. Soc. 2000, 122, 10244-5.
34. Suga, S.; Yamada, D.; Yoshida, J. Cationic three-component coupling involving an optically active enamine derivative. from time integration to space integration of reactions. Chem. Lett. 2010, 39, 404-6.
35. Sezer, N.; Bayhan, S.; Fesli, U.; Sanfilippo, A. A comprehensive review of the state-of-the-art of proton exchange membrane water electrolysis. Mater. Sci. Energy. Technol. 2025, 8, 44-65.
36. Xing, J.; Zeng, Z.; Best, W.; et al. Long-term durability test of highly efficient membrane electrode assemblies for anion exchange membrane seawater electrolyzers. J. Power. Sources. 2023, 558, 232564.
37. Brachi, M.; El Housseini, W.; Beaver, K.; et al. Advanced electroanalysis for electrosynthesis. ACS. Org. Inorg. Au. 2024, 4, 141-87.
39. Sharma, S. Electro-organic reactions: direct and indirect electrolysis. Orient. J. Chem. 2024, 40, 321-32.
40. Stuart, D. R.; Fagnou, K. The catalytic cross-coupling of unactivated arenes. Science 2007, 316, 1172-5.
41. Dohi, T.; Ito, M.; Morimoto, K.; Iwata, M.; Kita, Y. Oxidative cross-coupling of arenes induced by single-electron transfer leading to biaryls by use of organoiodine(III) oxidants. Angew. Chem. Int. Ed. Engl. 2008, 47, 1301-4.
42. Morofuji, T.; Shimizu, A.; Yoshida, J. Metal- and chemical-oxidant-free C-H/C-H cross-coupling of aromatic compounds: the use of radical-cation pools. Angew. Chem. Int. Ed. Engl. 2012, 51, 7259-62.
43. Fritz, H. P.; Ecker, P. Elektrochemische synthesen, XVIII: Reaktionen des Naphthalinradikalkations mit Nucleophilen. Chem. Ber. 1981, 114, 3643-54.
44. Chupakhin, O. N.; Charushin, V. N. Recent advances in the field of nucleophilic aromatic substitution of hydrogen. Tetrahedron. Lett. 2016, 57, 2665-72.
45. Gallardo, I.; Guirado, G. Electrochemical C–H functionalization of arenes and heteroarenes. In: Charushin V, Chupakhin O, editers. Metal free C–H functionalization of aromatics. Topics in Heterocyclic Chemistry. Springer, Cham; 2013. pp. 241-75.
46. Chupakhin, O. N.; Shchepochkin, A. V.; Charushin, V. N. Atom- and step-economical nucleophilic arylation of azaaromatics via electrochemical oxidative cross C–C coupling reactions. Green. Chem. 2017, 19, 2931-5.
47. Sun, G. Q.; Yu, P.; Zhang, W.; et al. Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations. Nature 2023, 615, 67-72.
48. Zhong, G.; Huang, Y.; He, L. Regioselectivity of N-heteroarene electrocarboxylations: divided vs. undivided cell. Chem. Synth. 2023, 3, 18.
49. Langlois, B. R.; Laurent, E.; Roidot, N. Trifluoromethylation of aromatic compounds with sodium trifluoromethanesulfinate under oxidative conditions. Tetrahedron. Lett. 1991, 32, 7525-8.
50. Ji, Y.; Brueckl, T.; Baxter, R. D.; et al. Innate C-H trifluoromethylation of heterocycles. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14411-5.
51. O’Brien, A. G.; Maruyama, A.; Inokuma, Y.; Fujita, M.; Baran, P. S.; Blackmond, D. G. Radical C-H functionalization of heteroarenes under electrochemical control. Angew. Chem. Int. Ed. Engl. 2014, 53, 11868-71.
52. Tommasino, J. B.; Brondex, A.; Médebielle, M.; Thomalla, M.; Langlois, B. R.; Billard, T. Trifluoromethylation reactions with potassium trifluoromethanesulfinate under electrochemical oxidation. Synlett 2002, 10, 1697-9.
53. Lu, M. C.; Shao, H. L.; Liu, T.; You, Q. D.; Jiang, Z. Y. Discovery of 2-oxy-2-phenylacetic acid substituted naphthalene sulfonamide derivatives as potent KEAP1-NRF2 protein-protein interaction inhibitors for inflammatory conditions. Eur. J. Med. Chem. 2020, 207, 112734.
54. Xu, F.; Jia, Y.; Wen, Q.; et al. Synthesis and biological evaluation of N-(4-hydroxy-3-mercaptonaphthalen-1-yl)amides as inhibitors of angiogenesis and tumor growth. Eur. J. Med. Chem. 2013, 64, 377-88.
55. Xu, F.; Xu, H.; Wang, X.; et al. Discovery of N-(3-((7H-purin-6-yl)thio)-4-hydroxynaphthalen-1-yl)-sulfonamide derivatives as novel protein kinase and angiogenesis inhibitors for the treatment of cancer: synthesis and biological evaluation. Part III. Bioorg. Med. Chem. 2014, 22, 1487-95.
56. Röhrig, U. F.; Awad, L.; Grosdidier, A.; et al. Rational design of indoleamine 2,3-dioxygenase inhibitors. J. Med. Chem. 2010, 53, 1172-89.
57. Cee, V. J.; Cheng, A. C.; Romero, K.; et al. Pyridyl-pyrimidine benzimidazole derivatives as potent, selective, and orally bioavailable inhibitors of Tie-2 kinase. Bioorg. Med. Chem. Lett. 2009, 19, 424-7.
58. Kise, N.; Manto, T.; Sakurai, T. Electroreductive coupling of phthalimides with α,β-unsaturated carbonyl compounds and subsequent acid-catalyzed rearrangement to 4-aminonaphthalen-1-ols: density functional theory study of the acid-catalyzed rearrangement of ketene silyl acetals. J. Org. Chem. 2021, 86, 18232-46.
59. Kise, N.; Isemoto, S.; Sakurai, T. Electroreductive coupling of phthalimides with alpha,beta-unsaturated esters: unusual rearrangement of resulting silyl ketene acetals. Org. Lett. 2009, 11, 4902-5.
60. Amines: synthesis, properties and applications By S. A. Lawrence. Cambridge University Press: Cambridge. 2004. 371 + x pp. £100/$180. ISBN 0-521-78284-8. Org. Process. Res. Dev. 2005, 9, 1016.
61. Wang, S.; Hanna, D.; Sugamori, K. S.; Grant, D. M. Primary aromatic amines and cancer: novel mechanistic insights using 4-aminobiphenyl as a model carcinogen. Pharmacol. Ther. 2019, 200, 179-89.
62. Wienhöfer, G.; Sorribes, I.; Boddien, A.; et al. General and selective iron-catalyzed transfer hydrogenation of nitroarenes without base. J. Am. Chem. Soc. 2011, 133, 12875-9.
63. Morofuji, T.; Shimizu, A.; Yoshida, J. Electrochemical C-H amination: synthesis of aromatic primary amines via N-arylpyridinium ions. J. Am. Chem. Soc. 2013, 135, 5000-3.
64. Brotzel, F.; Kempf, B.; Singer, T.; Zipse, H.; Mayr, H. Nucleophilicities and carbon basicities of pyridines. Chemistry 2007, 13, 336-45.
65. Li, Y.; Asaoka, S.; Yamagishi, T.; Iyoda, T. Electrochemical synthesis of pyridinium-conjugated assembly based on nucleophilic substitution of pyrene/perylene π-radical cation. Electrochemistry 2004, 72, 171-4.
66. Sharma, P. C.; Sinhmar, A.; Sharma, A.; Rajak, H.; Pathak, D. P. Medicinal significance of benzothiazole scaffold: an insight view. J. Enzyme. Inhib. Med. Chem. 2013, 28, 240-66.
67. Yeh, V.; Iyengar, R. Oxazoles. Comprehensive heterocyclic chemistry III. Elsevier; 2008. pp. 487-543.
68. Morofuji, T.; Shimizu, A.; Yoshida, J. Electrochemical intramolecular C-H amination: synthesis of benzoxazoles and benzothiazoles. Chemistry 2015, 21, 3211-4.
69. Zhang, G.; Liu, C.; Yi, H.; et al. External oxidant-free oxidative cross-coupling: a photoredox cobalt-catalyzed aromatic C-H thiolation for constructing C-S bonds. J. Am. Chem. Soc. 2015, 137, 9273-80.
70. Guru, M. M.; Ali, M. A.; Punniyamurthy, T. Copper(II)-catalyzed conversion of bisaryloxime ethers to 2-arylbenzoxazoles via C-H functionalization/C-N/C-O bonds formation. Org. Lett. 2011, 13, 1194-7.
71. Wesenberg, L. J.; Herold, S.; Shimizu, A.; Yoshida, J. I.; Waldvogel, S. R. New approach to 1,4-benzoxazin-3-ones by electrochemical C-H amination. Chemistry 2017, 23, 12096-9.
72. Brahma, K.; Das, B.; Chowdhury, C. A palladium-catalyzed facile and general method for the stereoselective synthesis of (E)-3-arylidene-3,4-dihydro-2H-1,4-benzoxazines and their naphthoxazine analogues. Tetrahedron 2014, 70, 5863-71.
73. Kononov, A. I.; Strekalova, S. O.; Morozov, V. I.; et al. Replacing sulfuric acid with water in electrochemical metal-free mild aromatic C–H amidation: a direct route to N-phenylamides. Org. Chem. Front. 2024, 11, 5820-30.
74. Fu, Y.; Zhang, L.; Sun, M.; et al. Direct electrochemical ritter-type amination of electron-deficient arenes. Eur. J. Org. Chem. 2023, 26, e202300553.
75. Moragas, T.; Correa, A.; Martin, R. Metal-catalyzed reductive coupling reactions of organic halides with carbonyl-type compounds. Chemistry 2014, 20, 8242-58.
76. Cheng, L. J.; Mankad, N. P. Copper-catalyzed carbonylative coupling of alkyl halides. Acc. Chem. Res. 2021, 54, 2261-74.
77. Knappke, C. E.; von Wangelin, A. J. 35 years of palladium-catalyzed cross-coupling with Grignard reagents: how far have we come? Chem. Soc. Rev. 2011, 40, 4948-62.
78. Dong, C. P.; Nakamura, K.; Taniguchi, T.; et al. Synthesis of aryl iodides from arylhydrazines and iodine. ACS. Omega. 2018, 3, 9814-21.
79. Ilangovan, A.; Satish, G. Direct amidation of 2’-aminoacetophenones using I2-TBHP: a unimolecular domino approach toward isatin and iodoisatin. J. Org. Chem. 2014, 79, 4984-91.
80. Dahiya, A.; Sahoo, A. K.; Chakraborty, N.; Das, B.; Patel, B. K. Updates on hypervalent-iodine reagents: metal-free functionalisation of alkenes, alkynes and heterocycles. Org. Biomol. Chem. 2022, 20, 2005-27.
81. Sano, K.; Kimura, N.; Kochi, T.; Kakiuchi, F. Palladium-catalyzed C−H iodination of N-(8-quinolinyl)benzamide derivatives under electrochemical and non-electrochemical conditions. Asian. J. Org. Chem. 2018, 7, 1311-4.
82. Möckel, R.; Hille, J.; Winterling, E.; Weidemüller, S.; Faber, T. M.; Hilt, G. Electrochemical synthesis of aryl iodides by anodic iododesilylation. Angew. Chem. Int. Ed. Engl. 2018, 57, 442-5.
83. Weissman, S. A.; Anderson, N. G. Design of experiments (DoE) and process optimization. a review of recent publications. Org. Process. Res. Dev. 2015, 19, 1605-33.
84. Murray, P. M.; Bellany, F.; Benhamou, L.; Bučar, D. K.; Tabor, A. B.; Sheppard, T. D. The application of design of experiments (DoE) reaction optimisation and solvent selection in the development of new synthetic chemistry. Org. Biomol. Chem. 2016, 14, 2373-84.
85. Gensch, T.; Glorius, F. Synthetic chemistry. The straight dope on the scope of chemical reactions. Science 2016, 352, 294-5.
86. Kataoka, K.; Hagiwara, Y.; Midorikawa, K.; Suga, S.; Yoshida, J. Practical electrochemical iodination of aromatic compounds. Org. Process. Res. Dev. 2008, 12, 1130-6.
87. Midorikawa, K.; Suga, S.; Yoshida, J. Selective monoiodination of aromatic compounds with electrochemically generated I+ using micromixing. Chem. Commun. 2006, 3794-6.
88. Yan, L.; Lei, H.; Yang, P.; Zhang, W. Electrochemically generated iodine cations from a glassy carbon electrode for highly selective iodination of anisole. Trans. Tianjin. Univ. 2022, 28, 433-9.
89. Shirakawa, E.; Hayashi, Y.; Itoh, K.; et al. Cross-coupling of aryl Grignard reagents with aryl iodides and bromides through SRN1 pathway. Angew. Chem. Int. Ed. Engl. 2012, 51, 218-21.
90. Schilz, M.; Plenio, H. A guide to Sonogashira cross-coupling reactions: the influence of substituents in aryl bromides, acetylenes, and phosphines. J. Org. Chem. 2012, 77, 2798-807.
91. Yang, Q. L.; Wang, X. Y.; Wang, T. L.; et al. Palladium-catalyzed electrochemical C-H bromination using NH4Br as the brominating reagent. Org. Lett. 2019, 21, 2645-9.
92. Zhang, P.; Sheng, X.; Chen, X.; et al. Paired electrocatalytic oxygenation and hydrogenation of organic substrates with water as the oxygen and hydrogen source. Angew. Chem. Int. Ed. Engl. 2019, 58, 9155-9.
93. Sbei, N.; Hardwick, T.; Ahmed, N. Green chemistry: electrochemical organic transformations via paired electrolysis. ACS. Sustainable. Chem. Eng. 2021, 9, 6148-69.
94. Chen, H.; Shen, C.; Dong, K. Parallel paired photoelectrochemical bromination of alkylarenes with electrochemical pinacol coupling. J. Org. Chem. 2024, 89, 2550-5.
95. Fry, A. J.; Reed, R. G. Electrochemical reduction of stereoisomeric geminal dihalonorbornanes. J. Am. Chem. Soc. 1972, 94, 8475-84.
96. Ikeda, S.; Tsukamoto, G.; Nishiguchi, I.; Hirashima, T. A novel synthetic method for penicillanic acid derivatives by electroreduction. Chem. Pharm. Bull. 1988, 36, 1976-81.
97. Kargin, Y. M.; Gritsenko, E. I.; Yanilkin, V. V.; et al. Electrochemical reduction of 1,1-dihalo-2,2-disubstituted cyclopropanes. Russ. Chem. Bull. 1992, 41, 1572-9.
98. Walborsky, H. M.; Hamdouchi, C. The nature of electron transfer from metal surfaces to the carbon-halogen bond. J. Am. Chem. Soc. 1993, 115, 6406-8.
99. Webb, J. L.; Mann, C. K.; Walborsky, H. M. Cyclopropanes. XXVI. Electrolytic reduction of optically active 1-halo-1-methyl-2,2-diphenylcyclopropanes. J. Am. Chem. Soc. 1970, 92, 2042-51.
100. Jaouannet, S.; Hazard, R.; Tallec, A. Electroreduction of optically active 1-bromo 1-carbomethoxy-2,2-diphenylcyclopropane: Factors influencing stereoselectivity. J. Electroanal. Chem. 1980, 111, 397-400.
101. Gütz, C.; Bänziger, M.; Bucher, C.; Galvão, T. R.; Waldvogel, S. R. Development and scale-up of the electrochemical dehalogenation for the synthesis of a key intermediate for NS5A inhibitors. Org. Process. Res. Dev. 2015, 19, 1428-33.
102. Fensterbank, L.; Goddard, J.; Malacria, M. 10.07 - C–C bond formation (Part 1) by addition reactions: through carbometallation catalyzed by group 8-11 metals. In: Comprehensive organometallic chemistry III. Elsevier; 2007. pp. 299-368.
103. Littke, A. F.; Fu, G. C. Palladium-catalyzed coupling reactions of aryl chlorides. Angew. Chem. Int. Ed. 2002, 41, 4176-211.
104. Konishi, M.; Tsuchida, K.; Sano, K.; Kochi, T.; Kakiuchi, F. Palladium-catalyzed ortho-selective C-H chlorination of benzamide derivatives under anodic oxidation conditions. J. Org. Chem. 2017, 82, 8716-24.
105. Ashikari, Y.; Nokami, T.; Yoshida, J. Integrated electrochemical-chemical oxidation mediated by alkoxysulfonium ions. J. Am. Chem. Soc. 2011, 133, 11840-3.
106. Phan, T. B.; Nolte, C.; Kobayashi, S.; Ofial, A. R.; Mayr, H. Can one predict changes from SN1 to SN2 mechanisms? J. Am. Chem. Soc. 2009, 131, 11392-401.
107. Adhami, W.; Richel, A.; Len, C. A review of recent advances in the production of furfural in batch system. Mol. Catal. 2023, 545, 113178.
108. Werpy, T.; Petersen, G. Top value added chemicals from biomass: Volume I - Results of screening for potential candidates from sugars and synthesis gas. 2004. https://www.nrel.gov/docs/fy04osti/35523.pdf. (accessed on 24 Mar 2025)
109. Cao, Y.; Knijff, J.; Delparish, A.; d’Angelo, M. F. N.; Noёl, T. A divergent paired electrochemical process for the conversion of furfural using a divided-cell flow microreactor. ChemSusChem 2021, 14, 590-4.
110. Wu, H.; Song, J.; Liu, H.; et al. An electrocatalytic route for transformation of biomass-derived furfural into 5-hydroxy-2(5H)-furanone. Chem. Sci. 2019, 10, 4692-8.
111. Cao, Y.; Noël, T. Efficient electrocatalytic reduction of furfural to furfuryl alcohol in a microchannel flow reactor. Org. Process. Res. Dev. 2019, 23, 403-8.
112. Wu, Y.; Liu, C.; Wang, C.; Lu, S.; Zhang, B. Selective transfer semihydrogenation of alkynes with H2O (D2O) as the H (D) source over a Pd-P cathode. Angew. Chem. Int. Ed. Engl. 2020, 59, 21170-5.
113. Kakiuchi, F.; Kochi, T. Transition-metal-catalyzed carbon-carbon bond formation via carbon-hydrogen bond cleavage. Synthesis 2008, 2008, 3013-39.
114. Saito, F.; Aiso, H.; Kochi, T.; Kakiuchi, F. Palladium-catalyzed regioselective homocoupling of arenes using anodic oxidation: formal electrolysis of aromatic carbon–hydrogen bonds. Organometallics 2014, 33, 6704-7.
115. Hull, K. L.; Lanni, E. L.; Sanford, M. S. Highly regioselective catalytic oxidative coupling reactions: synthetic and mechanistic investigations. J. Am. Chem. Soc. 2006, 128, 14047-9.
116. Zhu, Y.; Dong, W.; Tang, W. Palladium-catalyzed cross-couplings in the synthesis of agrochemicals. Adv. Agrochem. 2022, 1, 125-38.
117. Ma, C.; Zhao, C. Q.; Li, Y. Q.; et al. Palladium-catalyzed C-H activation/C-C cross-coupling reactions via electrochemistry. Chem. Commun. 2017, 53, 12189-92.
118. Chen, X.; Goodhue, C. E.; Yu, J. Q. Palladium-catalyzed alkylation of sp2 and sp3 C-H bonds with methylboroxine and alkylboronic acids: two distinct C-H activation pathways. J. Am. Chem. Soc. 2006, 128, 12634-5.
119. Engle, K. M.; Mei, T. S.; Wasa, M.; Yu, J. Q. Weak coordination as a powerful means for developing broadly useful C-H functionalization reactions. Acc. Chem. Res. 2012, 45, 788-802.
120. Li, Y. Q.; Yang, Q. L.; Fang, P.; Mei, T. S.; Zhang, D. Palladium-catalyzed C(sp2)-H acetoxylation via electrochemical oxidation. Org. Lett. 2017, 19, 2905-8.
121. Dick, A. R.; Hull, K. L.; Sanford, M. S. A highly selective catalytic method for the oxidative functionalization of C-H bonds. J. Am. Chem. Soc. 2004, 126, 2300-1.
122. Li, L. J.; Jiang, Y. Y.; Lam, C. M.; Zeng, C. C.; Hu, L. M.; Little, R. D. Aromatic C-H bond functionalization induced by electrochemically in situ generated tris(p-bromophenyl)aminium radical cation: cationic chain reactions of electron-rich aromatics with enamides. J. Org. Chem. 2015, 80, 11021-30.
123. See, Y. Y.; Herrmann, A. T.; Aihara, Y.; Baran, P. S. Scalable C-H oxidation with copper: synthesis of polyoxypregnanes. J. Am. Chem. Soc. 2015, 137, 13776-9.
124. Yang, Q. L.; Li, Y. Q.; Ma, C.; Fang, P.; Zhang, X. J.; Mei, T. S. Palladium-catalyzed C(sp3)-H oxygenation via electrochemical oxidation. J. Am. Chem. Soc. 2017, 139, 3293-8.
125. Robinson, S. G.; Mack, J. B. C.; Alektiar, S. N.; Du Bois, J.; Sigman, M. S. Electrochemical Ruthenium-catalyzed C-H hydroxylation of amine derivatives in aqueous acid. Org. Lett. 2020, 22, 7060-3.
126. Mack, J. B. C.; Gipson, J. D.; Du Bois, J.; Sigman, M. S. Ruthenium-catalyzed C-H hydroxylation in aqueous acid enables selective functionalization of amine derivatives. J. Am. Chem. Soc. 2017, 139, 9503-6.
127. Rebsdat, S.; Mayer, D. Ethylene Oxide. In: Ullmann’s Encyclopedia of Industrial Chemistry. Wiley; 2001. https://www.ugr.es/~tep028/pqi/descargas/Industria%20quimica%20organica/tema_5/oxido_etileno_a10_117.pdf. (accessed on 24 Mar 2025).
128. Rafiee, M.; Wang, F.; Hruszkewycz, D. P.; Stahl, S. S. N-hydroxyphthalimide-mediated electrochemical iodination of methylarenes and comparison to electron-transfer-initiated C-H functionalization. J. Am. Chem. Soc. 2018, 140, 22-5.
129. Xiang, H.; He, J.; Qian, W.; et al. Electroreductively induced radicals for organic synthesis. Molecules 2023, 28, 857.
130. Xu, H.; Yu, B.; Zhang, H.; et al. Reductive cleavage of inert aryl C-O bonds to produce arenes. Chem. Commun. 2015, 51, 12212-5.
131. Chernowsky, C. P.; Chmiel, A. F.; Wickens, Z. K. Electrochemical activation of diverse conventional photoredox catalysts induces potent photoreductant activity. Angew. Chem. Int. Ed. Engl. 2021, 60, 21418-25.
132. Fokin, I.; Siewert, I. Chemoselective electrochemical hydrogenation of ketones and aldehydes with a well-defined base-metal catalyst. Chemistry 2020, 26, 14137-43.
133. Hoque, M. A.; Gerken, J. B.; Stahl, S. S. Synthetic dioxygenase reactivity by pairing electrochemical oxygen reduction and water oxidation. Science 2024, 383, 173-8.
134. Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Recent progress in applications of enzymatic bioelectrocatalysis. Catalysts 2020, 10, 1413.
135. Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct electron transfer-type bioelectrocatalysis of redox enzymes at nanostructured electrodes. Catalysts 2020, 10, 236.
136. Francke, R.; Májek, M. Mediated electron transfer in electrosynthesis: concepts, applications, and recent influences from photoredox catalysis. In: Inagi S, editor. Sustainable and functional redox chemistry. The Royal Society of Chemistry; 2022. pp. 119-53.
137. Shi, L.; Dong, H.; Reguera, G.; et al. Extracellular electron transfer mechanisms between microorganisms and minerals. Nat. Rev. Microbiol. 2016, 14, 651-62.
138. Dong, H.; Cheng, J.; Li, H.; Yue, L.; Xia, R.; Zhou, J. Electron transfer from Geobacter sulfurreducens to mixed methanogens improved methane production with feedstock gases of H2 and CO2. Bioresour. Technol. 2022, 347, 126680.
139. Freguia, S.; Virdis, B.; Harnisch, F.; Keller, J. Bioelectrochemical systems: microbial versus enzymatic catalysis. Electrochim. Acta. 2012, 82, 165-74.
140. Radhika, D.; Shivakumar, A.; Kasai, D.; Koutavarapu, R.; Peera, S. Microbial electrolysis cell as a diverse technology: overview of prospective applications, advancements, and challenges. Energies 2022, 15, 2611.
141. Bensaid, S.; Ruggeri, B.; Saracco, G. Development of a photosynthetic microbial electrochemical cell (PMEC) reactor coupled with dark fermentation of organic wastes: medium term perspectives. Energies 2015, 8, 399-429.
142. Hassan, R. Y. A.; Febbraio, F.; Andreescu, S. Microbial electrochemical systems: principles, construction and biosensing applications. Sensors 2021, 21, 1279.
Cite This Article

How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright















Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].