


Mutant SRF and YAP synthetic modified mRNAs drive cardiomyocyte nuclear replication

Abstract
Introduction: Aging is associated with sarcopenia, myocyte loss, and dysfunction. The problem is compounded as the adult heart lacks the regenerative capacity to self-repair. Serum response factor’s (SRF’s) dual activity is essential for cell replication and heart cell differentiation. SRF interacts with cofactors, such as NKX2-5 and GATA4, which give cardiac-specific gene activity, and ETS factors such as ELK1 drive cell replication. Recently, the mutant YAP-5SA of the Hippo pathway was implicated in cardiomyocyte proliferation and growth.
Aim: We hypothesized that disruption of interactions of SRF with NKX2-5 and GATA4 would lead to dedifferentiation of cardiomyocytes to a proliferative stem cell state and complement YAP-5SA to generate undifferentiated cardiomyocytes in a more primitive replicative state.
Methods and results: To weaken SRF interactions with NKX2-5 and GATA4, alanine scanning mutations were generated across the SRF N-terminus of the MADS-box. One SRF mutant, SRF153(A3), was tested along with the YAP-5SA mutant, as degradable synthetic modified mRNAs (mmRNAs), in rat primary cardiomyocytes. To measure cell replication, adult cardiomyocytes were pulsed with alpha-EdU and then DAPI stained, while gene activity was assayed by RNA sequencing. To measure chromatin remodeling, Transposon 5 was used in ATAC sequencing. We observed that single and triple alanine substitutions of mutants centering over SRF-Lys154 essentially blocked myocyte differentiation, and NKX2-5 and GATA4 failed to stabilize mutated SRF DNA binding. Instead, many stem cell factors including NANOG and OCT4 were induced. SRF153(A3) does not recognize SRF response elements per ATAC sequencing and consequently induces stem cell factors such as NANOG and OCT4, cardiomyocyte dedifferentiation, and cell cycle reentry. SRF153(A3) and YAP5SA mmRNA led to alpha-EDU incorporation in ~35% of the cardiomyocytes. DIAPH 3, a marker of the contractile ring during anaphase, appeared between and around replicated nuclei in three-month-old adult mouse cardiac myocytes. The combination of these synthetic mRNA increased nuclei replication with the expression of origin of replication genes, while genes associated with cardiomyocyte differentiation were down-regulated. ATAC sequencing revealed SRF153(A3) and YAP5SA mmRNA-induced chromatin remodeling of cell cycle, spindle, and growth factor genes by additive and synergistic activities.
Conclusion: SRF153(A3) synthetic mmRNA and the mutant YAP-5SA mmRNA induced cardiomyocyte dedifferentiation, to nuclear replication in adult cardiac myocytes. The combinatorial use of mmRNA encoding SRF153(A3) and YAP-5SA has the potential to become a powerful clinical strategy for treating human heart disease.

Keywords
INTRODUCTION
The loss of cardiomyocytes underlies most causes of heart failure, which is a major cause of mortality and morbidity in the elderly. In addition, aging is associated with loss of sarcomeres and cardiac myocyte dysfunction. Normal repair processes are inadequate to deal with extensive myocardial damage. While heart transplantation is the standard for treatment, the limited availability of donor hearts and the risk of rejection restrict its widespread use. The inescapable conclusion is that adult mammalian cardiomyocytes are refractory to mitotic activity, unlike those found in either early postnatal mice or zebrafish[1,2]. Interventions with in vitro-generated cardiomyocytes, produced from inducible pluripotent stem cells (iPSC) and/or embryonic stem (ES) cells, failed to produce the long-term repair. Recently, short-term in vivo transgene induction of reprogramming factors OCT3/4, SOX2, KLF4, and C-MYC (OKSM) for less than a week generated partial reprograming, rejuvenated senescent organs, and extended mouse lifespans[3]. Indeed, a recent study showed that in vivo expression of OKSM transgenes caused murine heart cell regeneration[4]. Short-term expression of OKSM did not cause cancer but was sufficient to induce cell replication and rejuvenation[4]. Thus, to rejuvenate senescent myocytes and expand their number after a cardiac infarct, adult myocytes may need to be taken backwards to a primitive replicative state driven by stem cell factors.
Serum response factor (SRF) is a member of an ancient DNA binding protein family that shares a highly conserved DNA-binding/dimerization domain of 90 amino acids termed the MADS box, as reviewed by Treisman[5] [Figure 1A]. MADS boxes have similar DNA binding specificities and dimerize to symmetrically contact the serum response element with a consensus sequence CC(A/T)6GG, also known as the SRE and/or CArG box[6]. A feature of a large number of cardiac and virtually all the smooth muscle-expressed genes to date is their dependence on CArG boxes[6]. The X-ray crystal structure of the SRF core bound to DNA[7], as shown in Figure 1B, revealed the coiled coil formed by the MADS box αI helices (Amino Acids 153-179), which lies parallel and on top of a narrow DNA major groove making contacts with the phosphate backbone on an SRE half-site. In addition, the unstructured N-terminal extension from the alpha I helix (Amino Acids 132-152) makes critical base contacts in the minor groove. The dimerization of the MADS box occurs above the alpha I helix by a structure composed of two beta-sheets in the monomer that interact with the same unit in its partner. A second alpha II helix in the C-terminal portion of the MADS box stacked above these beta-sheets completes this stratified structure, which in total provides contacts for binding co-accessory factors to SRF[7].

Figure 1. Serum response factor (SRF) association with fundamental cofactors. (A) Schematic diagram of the SRF regulatory domains including a highly conserved DNA-binding/dimerization domain of 90 amino acids termed the MADS box[4] and its transcriptional activation domain (TAD). (B) MADS box DNA contact sites and biophysical structures defined by X-ray crystal analysis[7] revealed the N-terminal extension (132-152 aa) that contacts the minor groove of the CArG box half site. Next, alpha I helices (153-179 aa) lie parallel and on top of a narrow DNA major groove and are followed by two beta-sheets separated by the beta-loop (190-199 aa), thus allowing for dimerization of the monomers. A second alpha II helix (204-224 aa) in the C-terminal portion of the MADS box completes the structure. (C) The STRING program (https://string-db.org/network/10090.ENSMUSP00000015749) identified the top 10 factors that have the strongest association with SRF to generate the network images for predicted associations with SRF (a node) and for cofactor proteins (second nodes). An edge may be drawn with up to seven differently colored lines which represent the existence of types of evidence used in predicting the associations: blue line (co-occurrence), purple line (experiments), yellow line (text mining), light blue line (database), and black line (co-expression). (D) The highest confidence scores are mostly above 0.9. These are the strongest enrichment scores supporting the importance of these proteins’ biological functional activity connected to SRF. (E) The schematic diagram of the SRF regulatory domains including the MADS box shows that the cofactors NKX2-5, GATA4/6, and CRP2 and inhibitor HOP1 bind to the N-terminal extension. ETS factors ELK1 and SAP1 bind to the beta loop and compete with MRTF-A. TEAD factors bind to the second alpha helix. YAP may indirectly bind to SRF through MRTF-A and TEAD interaction sites.
The appearance and diversification of nascent embryonic cardiac and smooth muscle cells require the combinatorial interactions of SRF with other transcription factors enriched in the early progenitor cells[8-10]. We showed that SRF is absolutely required for sarcomere formation and the first heartbeat in embryonic heart formation[10]. The STRING program identified the top 10 factors that have the strongest association with SRF, as shown in Figure 1C. STRING uses a spring model to generate the network images. The network view shown in Figure 1C summarizes predicted associations with SRF (a node) for cofactor proteins (second nodes) as follows: CREB1, ELK1, ELK4, ETS1, ETS2, GATA4, MKL1, MKL2, MYOCD, and NKX2-5[8-13]. The SRF network has significantly more interactions than expected, having more interactions among themselves than what would be expected for a random set of proteins of the same size and degree distribution drawn from the genome. Such strong enrichment scores, as shown in Figure 1D, indicate the importance of these proteins’ biological functional activity connected to SRF.
As shown in Figure 1E, many of these cofactors and others were mapped to their protein interactive sites in the MADS box. Mutations that prevent SRF binding severely impair the expression of c-fos[11,12], as well as muscle-restricted genes[8-10]. An ETS factor, such as ELK1, has an interactive B box that stabilizes SRF binding to DNA, especially in the presence of an adjacent ETS site[11,12]. In addition, CArG boxes recruit SRF and cofactors, such as NKX2-5[8], and GATA4[9,10], that strongly enhance SRF-DNA binding affinity, thus, permitting the formation of higher-order DNA binding complexes at relatively low SRF levels for cardiogenic specific promoters. Similarly, CArG boxes in smooth muscle gene promoters recruit the cysteine-rich 2 LIM protein, which bridges SRF and GATA6 factors through interaction with the MADS N-terminal extension[14], and MYOCARDIN, which competes with ETS factors that interact with the beta loop region of the MADS box[13], as shown in Figure 1. Other mediators of SRF-dependent activity are MAL or MKL[15-19], which provide the link between RhoA-dependent cytoskeletal regulation and SRF-dependent gene expression[15,18]. TEF1, renamed TEAD, also directly binds to the second alpha coil in the MADS box[20,21]. All of these cofactors greatly enhance SRF transactivation, dependent on the context of the promoter sequences.
We considered whether disruption of NKX2-5 and GATA4 co-interactions with SRF block cardiac differentiation gene programs. Thus, experiments were planned to first generate alanine scanning mutations across the SRF N-terminus up to the alpha I helix of the MADS box that interacts with NKX2-5 and GATA4. Figure 1E shows a schematic diagram of the MADS box domain binding to DNA. We used site-directed PCR mutagenesis to create amino acid substituted mutations at specified residues across the SRF N-terminus. Residues were changed to alanines (giving a neutral charge and removing potential interacting side chains). Our experimental design used SRF null murine ES cells rescued with lentiviral expression vectors driving an SRF mutant. This is important, because SRF mutant activity is not due to being mixed with endogenous SRF molecules. We identified an SRF mutant, named SRF153(A3), that no longer recognizes CArG boxes, nor is influenced by NKX2-5 and GATA4 interactions. Thus, we showed SRF153(A3) functions as a new synthetic transfer factor that blocks cardiac differentiation and sarcomerogenesis.
Could SRF153(A3) be drawn to other DNA binding targets that activate cell proliferation? An excellent example is the ETS factors, as previously mentioned[13]. Other mediators of SRF cell signaling, such as MAL or MKL[15-19], provide the link between RhoA-dependent cytoskeletal regulation and SRF-dependent gene expression[15,18]. TEAD, a critical YAP cofactor, also associates with MRTF-A[18] overlapping the MYOCARDIN binding site on the SRF’s MADS box leading to rho kinase activation and cell replication. We showed that TEAD also directly binds to the MADS box independent of MRTF-A[20,21].
Recently, manipulating the Hippo pathway has attracted interest as a strategy for increasing cardiac regeneration. Cardiac-specific knockouts of Sav1, Lats1/2, and Nf2 genes in mature cardiomyocytes revealed enhanced cardiomyocyte proliferation, and reduced scar formation post-MI[22,23]. Human YAP1 contains five phosphorylation HXRXXS motifs. Bin Zhao et al.[24] mutated YAP by replacing individual serine residues in the HXRXXS motifs with alanine, generating a YAP5SA mutant that resists phosphorylation. Central to the Hippo pathway is a cascade of phosphorylation events in which phosphorylation of YAP1 prevents shuttling of YAP1 into the nucleus and promotes 14-3-3 binding and protein degradation[24]. When the Hippo pathway is inactivated, unphosphorylated YAP1 enters the nucleus and binds to multiple transcription factors (e.g., TEAD/TEF1 and MRTF-A). YAP1 binding to its partners, such as TEAD and MRTF-A in the nucleus, typically promotes gene expression programs that favor proliferation. Even though YAP and SRF do not interact directly[25], we considered whether an SRF mutant SRF153(A3) which drives stem cell factors and causes cardiac myocytes to dedifferentiate would complement mutant YAP5SA to proliferate dedifferentiated myocytes.
METHODS
Alanine scanning mutagenesis
Identifying the sites of interaction with NKX2-5 and GATA4 in the region where SRF binds to the SRE is essential to unravel the mechanism of SRF’s transcriptional regulation of proliferation and/or cardiac-specific genes. All DNA contacts occur within the N-terminal portion of the MADS box, and SRF-SRF dimerization is mediated by elements within the MADS box together with additional residues from the immediately adjacent C terminal region[7]. SRF’s interactions with NKX2-5 and GATA4 map to Amino Acids 142-171 in the MADS box. NKX2-5 and GATA4 markedly increase both SRF binding to the cardiac
Plasmid construction
Full-length cDNA of SRF and YAP was amplified by PCR and cloned into indicated vectors including pRK7-N-FLAG. Point mutations for SRF were generated by site-directed mutagenesis.
Synthesis of mRNA
SRF, YAP, SRF153(A3), and YAP5SA mRNAs were transcribed in vitro from linearized T7 plasmid templates using mMESSAGE mMACHINE T7 Transcription Kit (ThermoFisher). In vitro transcription reaction was conducted at 37 °C overnight. Lithium chloride (LiCl) precipitation was conducted at -20 °C for 72 h to remove unincorporated nucleotides and most proteins. Purified mRNAs were quantified by NanoDrop Microvolume Spectrophotometers (ThermoFisher) and analyzed by agarose gel electrophoresis. Then, 1 µg mRNA was loaded to the gel, and clear bands were observed before mRNAs were tested in cells.
Cell culture conditions
NIH/3T3 (mouse fibroblast) cell line was from the University of Houston’s Biology and Biochemistry research laboratory. NIH/3T3 cells were cultured in DMEM (Gibco) supplemented with 10% (vol/vol) heat-activated FBS (GenDEPOT) and 100 U/mL penicillin. Rat cardiomyocytes (RCm) (Cell Applications) were primary cells derived from normal neonatal rat heart tissue and plated on plates immediately after primary isolation. RCms were cultured in RCm Culture Medium (Cell Applications) with growth supplements (Cell Applications). NIH/3T3 and Rat cardiomyocyte cultures were maintained at 37 °C in a humidified atmosphere containing 5% (vol/vol) CO2. Adult mouse cardiomyocytes were primary cells isolated from adult mouse heart tissue. Adult mouse cardiomyocytes were cultured in plating medium (100 U/mL penicillin, 2 mM glutamine, 10% calf serum, 10 mM BDM, and 2 mM ATP in MEM) for 1 h after plating, and then changed to culture medium (100 U/mL penicillin, 2 mM glutamine, 1 mg/mL BSA, 10 mM BDM, and 0.005 ug/mL ITS in MEM) for long-term culture.
Cell transfection
Cell mRNA transfection was carried out by Lipofectamine MessengerMAX (ThermoFisher). First, 500 ng mRNA were added to 50 µL Opti-MEM medium (ThermoFisher) with 1.5 µL Lipofectamine MessengerMAX reagent and incubated for 15 min before adding to the cells in 24-well plates. The culture medium was changed 6 h after transfection.
Quantitative PCR
Cells were scratched from plates and total RNA was isolated with Zymo or Millipore total RNA extraction kit. RNA (1 µg) was used per reaction to make cDNA with QuantaBio cDNA mix. The mix was incubated in an Applied Biosystem 7500 qPCR machine with the program 7500 fast reaction, and quantitative standard curves were measured using standard ramp speed. The cycle was initiated at 48 °C for 10 min and 95 °C for 3 min, and then 40 cycles of 95 °C for 10 s and 60 °C for 1 min were used for detection of relative mRNA expression of desired rodent genes. Relative levels of RNA transcripts were the following: NANOG was detected with the forward primer (5’-3’) TCATTTGTCGGCCTGAACAAAA and reverse primer (5’-3’) GAGGCATCTCAGCAGAA GACA; OCT4 was detected with the forward primer (5’-3’) CTTGAATCCCGAATGG AAAGGG and reverse primer (5’-3’) CCTTCCCAAATAGAACCCCCA; KLF4 was detected with the forward primer (5’-3’) AAGCAGGAGGCGGTCTCTT and reverse primer (5’-3’) CCGGGACTGACCTTGGTAA; and SRF and mutants were detected with the forward primer (5’-3’) CAGACCCCACAACAGACCAG and reverse primer (5’-3’) CTGACTTGCATGGTGG TAGAG.
Immunofluorescence staining
Cells were seeded in a 24-well plate with a #1.5 coverslip at the bottom of the well and treated with the reagents indicated. Cells were washed in phosphate buffered saline (PBS); fixed in 4% (wt/vol) PFA; immuno-stained overnight with anti-SRF (G-20, Santa Cruz Biotechnology, 1/200), anti-YAP (ThermoFisher, 1/200), anti-TNNT (Abcam, 1/200), anti-TUBULIN antibody (CST, 1/200) with 1% BSA (Sigma Aldrich), anti-DIAPH3 (ThermoFisher, 1/200), and anti-NANOG (Sigma Life Science, 1/200); and washed three times with PBS followed by a 1 h exposure to a FITC-conjugated/TRITC-conjugated secondary antibody (ThermoFisher). Slides were mounted with ProLong Diamond Antifade Mountant (ThermoFisher). Immunofluorescence was visualized by a Leica SP8 confocal microscope.
Western blot
Cells were washed twice with PBS before being scraped and pelleted. NP-40 lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 1 mg/mL aprotinin, 1 mg/mL leupeptin, 1 mg/mL pepstatin, 1 mM Na3VO4, and 1 mM phenylmethylsulfonylfluoride) was used to resuspend the cells and lyse the cells for 30 min. The cell lysis was centrifuged at 12,000 g for 15 min at 4 °C, and the insoluble fraction was discarded after centrifuge. The supernatant was fractionated by SDS/PAGE gel in Bolt MES SDS Running Buffer (ThermoFisher) and transferred to nitrocellulose filter membranes in NuPAGE transfer buffer (ThermoFisher). The membranes were blocked with 5% nonfat dry milk at room temperature for 1 h and then incubated with antibodies against SRF, YAP, and/or NANOG overnight at 4 °C. The membranes were washed three times with Tris-buffered saline with Tween 20 (TBST) before incubation with secondary antibodies.
EdU assay
Alpha-EdU solution was added to 24-well plates to make a final concentration of 10 µM. EdU was pulsed for 8 h in neonatal cardiomyocyte and fibroblast experiments. In adult cardiomyocyte experiments, alpha-EdU was pulsed for 4 h. Alpha-EdU was detected by Click-iT EdU Cell Proliferation Kit (ThermoFisher).
Cell counting
DAPI and alpha-EdU positive cells were counted by ImageJ software (NIH).
RNA sequencing and analysis
Rat neonatal cardiomyocytes were harvested 32, 40, and 48 h after individual treatment of SRF153(A3) mmRNA, YAP5SA mmRNA, and a combination of both SRF153(A3) and YAP5SA mmRNA delivered with Lipofectamine MessengerMAX (ThermoFisher). RNA was extracted using miRNeasy Mini Kit (Qiagen) with on-column RNase-Free DNase (Qiagen) digestion following the manufacturer’s instructions. Extracted RNA samples underwent quality control assessment using the RNA tape on Tapestation 4200 (Agilent) and were quantified with Qubit Fluorometer (ThermoFisher). Kallisto was used for pseudo alignment and transcript quantification[26]. Differential gene expression quantification was performed using DESeq2 software according to the manufacturer’s instructions[27]. Differentially expressed genes (DEG) were determined with fold change > 1.5 and P-FDR-adjusted < 0.1 genes (FDR: false discovery rate) with more than one reads per kilobase of the transcript, per million mapped reads (RPKM), under the test conditions as compared to the controls. Gene set enrichment analysis (GSEA) was conducted using javaGSEA2-3.0[28]. Gene ontology analysis was performed using Bioconductor-clusterProfiler packages[29]. All tables and figures in the RNA-seq analysis were plotted by R-studio (3.6.0). Heat maps were plotted using the R “pheat map” package[30].
ATAC-seq and analysis
Rat neonatal cardiomyocytes were harvested 24 h after individual treatments of SRF mmRNA, YAP mmRNA, SRF153(A3) mmRNA, YAP5SA mmRNA, and a combination of both SRF153(A3) and YAP5SA mmRNA delivered by Lipofectamine MessengerMAX (ThermoFisher). Around 300,000 cells for each sample were submitted for sequencing. Transfected neonatal cardiomyocytes were centrifuged at 500 × g at 4 °C and resuspended 500 µL of ice-cold cryopreservation solution (50% FBS, 40% cardiomyocyte growth media, 10% DMSO). Next, 500 µL of cell suspension for each sample were transferred to a 2 mL cryotube on ice. The cryotubes were then transferred to a pre-chilled Mr. Frosty container and placed at -80 °C overnight to complete cryopreservation. Approximately 300,000 cells were shipped frozen to Active Motif to perform Assay for Transposase-Accessible Chromatin using sequencing. The cells were thawed in a 37 °C water bath, pelleted, washed with cold PBS, and tagmented. Cell pellets were resuspended and pelleted in lysis buffer, and then tagmented using the enzyme and buffer (provided in the Illumina Nextera Library Prep Kit). Tagmented DNA was then purified by the MinElute PCR purification kit (Qiagen), amplified with 10 cycles of PCR, and purified by using Agencourt AMPure SPRI beads (Beckman Coulter). The resulting material was quantified by the KAPA Library Quantification Kit for Illumina platforms (KAPA Biosystems) and sequenced with PE42 sequencing on the NextSeq 500 sequencer (Illumina).
Cutadapt (3.0) was used to remove the adapter and low-quality sequence from sequencing reads. Cleaned reads were then mapped to rn4 rat genome reference by Bowtie2 (2.2.7), with the “very-sensitive” option and “-k” being set as 10. After alignment, the narrow peaks were called by using MACS2 (2.2.6) with
Animals
C57BL/6J of both genders were housed and studied in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (eighth edition)[31]. Animals were handled according to Institutional Animal Care and Use Committee (IACUC) protocols
Microscopy
Brightfield and immunofluorescence images were obtained using a Nikon Ti-E inverted microscope equipped with a DS-Fi1 5-megapixel color camera (Nikon Instruments). Confocal images were obtained using a Leica SP8 Upright Confocal DM6000 CFS Microscope (Leica Microsystems).
RESULTS
SRF mutants drive the expression of stem cell marker genes and block cardiomyocyte-specific genes
The idea that SRF activity is largely controlled by its interaction with cofactors such as NKX2-5, GATA4, and others was tested by a “gain-of-function” approach applied to SRF null ES cells through the lentiviral rescue of murine wildtype ES cells AB2.2 and SRF−/− ES cells, as shown in Figure 2A[32]. In addition, cofactor gene expression by the lentiviral rescue of SRF null ES cells served as a screening tool to evaluate cofactors’ functional relationships with SRF. Null SRF ES cells were maintained at optimal conditions, as described in the Methods Section. Since NKX2-5 and GATA4 facilitate SRF-dependent activation of cardiac differentiated gene programs, would disruption of their co-interactions with SRF act as a default program to block differentiation? Human SRF cDNA and all the alanine substitution mutants with N terminal HA tag were cloned into the EcoRI site of the doxycycline-inducible lentiviral vector, which is rTA-responsive with Ptet-LTR1 promoter, in which we added a constitutive IRES-GFP tag [Figure 2B and C]. Infected ES cells were FACS enriched twice to augment the virally infected population to greater than 90% purity [Figure 2D]. Then, doxycycline was added to culture media to induce viral gene expression of SRFwt and SRF mutants, and RNA was isolated within two days after the plating of embryoid bodies. Figure 2B shows the triple alanine substitution mutations generated by PCR mutagenesis, as described previously[10], to generate scanning alanines starting at the beginning of the N-terminal extension to the beginning of the first alpha helix. Triple alanine mutations, induced by doxycycline in null SRF ES cells, were tested by electrophoretic mobility assays (EMSA) with a [P32]-labeled cardiac alpha-actin promoter DNA in the presence of lentiviral co-expressed NKX2-5 and/or GATA4 [Figure 2E]. As shown by EMSA, GATA4 and NKX2-5 enhanced the binding of wild-type SRF. In comparison, the triple alanine mutations starting at 141, 147, and/or 150 poorly bound DNA, and their binding activity was not facilitated by the presence of co-expressed NKX2-5 and GATA4, even though NKX2-5 showed considerable direct DNA binding to an NKX site embedded within the second and third CArG boxes[9]. Note that there are no GATA binding sites within the alpha cardiac actin promoter[9]. The SRF153(A3) mutant displayed weak EMSA binding that was not improved by GATA4 and/or NKX2-5. Thus, SRF mutants that failed to shift with NKX2-5 and GATA4 in EMSA also failed to activate cardiac actin promoter-luciferase reporter activity [Figure 2F].

Figure 2. Rescue of SRF null ES cells with virally expressed SRFwt and SRF triple alanine substitution mutants reveal critical SRF contacts for NKX2-5 and Gata4. To quantitate and isolate reprogrammed SRF null ES cells, we used a FACS strategy in which lentiviral infection of SRF null ES with SRF mutants was detected by expression of lentiviral expressed IRES-GFP. (A) Schematic diagram of our lentiviral infection transduction protocol into freshly trypsin-dissociated SRF null ES cells mixed with lentivirus at a multiplicity of infection of 100 using 8 μg/mL polybrene (Sigma), containing doxycycline-induced control virus (rTTA), SRF, and other SRF(A3) scanning mutants. (B) Alanine scanning mutations (shown as the letter a) were generated by PCR mutagenesis from Amino Acids 141 to 155 in the MADS Box. (C) Schematic diagram of the full-length cDNA of SRF and SRF(A3) mutants were amplified by PCR and cloned into the doxycycline lentiviral vector rTA-responsive vector with Ptet-LTR1 promoter and the T7 plasmid vector. (D) Three days later, the upper 50% of EGFP+ cells were twice enriched and isolated by FACS (Beckman-Coulter Altra) cultured under growth conditions and FACS sorted again for further enrichment to approximately 90%. Enriched infected ES cells were suspended in hanging drops on Day 5 and then treated with Doxycycline for the next three days, which induced expression of SRF and SRF(A3) mutants, as evaluated by protein blots. On Day 7, cellular aggregates were collected and plated to Day 8 and collected for analysis. (E) Extracts of SRF null murine ES cells rescued by lentiviral expressed SRF(A3) mutants, spanning the N-terminal extension to the start of alpha-helix 1, were used in EMSA DNA binding assays with the [P32]-labeled alpha-cardiac promoter in the presence of lentiviral co-expressed NKX2-5 and/or GATA4. (F) Luciferase reporter assay of viral transfected alpha cardiac actin promoter luciferase reporter with SRF mutants and virally co-expressed NKX2-5 and/or GATA4. SRF: Serum response factor; ES: embryonic stem.
Next, we dissected key alanine substitutions by their predicted DNA contact sites, as shown by X-ray crystal analysis[7] [Figure 3A] and validated by EMSA DNA binding [Figure 2E]. We scrutinized the X-ray crystal analysis of the SRF complex with a specific CArG[7], which showed that amino acid substitution at Lys154 was the most critical residue for DNA binding between Amino Acid 153 and 155 [Figure 3A]. A computational program that predicts the effects of mutations in protein coding regions on nucleic acid binding affinities[33] showed the predicted G at Lys154 was -1.996, stronger than for N153 and/or L155 [Figure 3B]. The alanine substitution at N153A and L155A did not block DNA binding or facilitate SRF’s EMSA with NKX2-5 and/or GATA4 [Figure 3C]. The single point mutation at K154A efficiently blocked DNA binding, which also could not be stabilized by NKX2-5 and GATA4. ELK1, an ETS factor containing a B box, stabilized and facilitated the binding of the SRF153(A3) and the SRF mutant K154A by binding to the c-fos promoter, which has an ETS binding site adjacent to the SRF binding CArG box[12] [Figure 3D]. In contrast, the cardiac alpha-actin promoter does not contain ETS sites adjacent to CArG boxes. Thus, depending on the target context, the SRF mutant SRF153(A3) would likely block cardiogenic specific genes that are dependent on NKX2-5 and GATA4 co-association. Analysis of SRF153(A3) binding targets by ATAC sequencing revealed the reduced ability of SRF153(A3) to bind to consensus CArG sequences. Instead, SRF153(A3) may depend on tethering with other transcription factors, such as ETS factors, to bind to DNA targets other than CArG sequences (see Table 1, ATAC sequencing targets).

Figure 3. SRF153(A3) mutant facilitates cell replication but not myocyte differentiation in the rescue of lentiviral infected SRF null murine ES cells. (A) A reproduction drawing of the key MADS box DNA contacts is shown[7]. Note that alanine substitution at Lys154 was the most critical residue for DNA binding between Amino Acids 153 and 155. (B) A computational program[27] predicted ΔΔG at Lys154 was -1.996, stronger than for N153 and/or L155. (C) EMSA of lentiviral expressed NKX2-5 and/or Gata4 in the context of the alpha cardiac actin promoter failed to stabilize the single alanine point mutant, Lys154Ala, a critical base contact. (D) EMSA of expressed ETS factor, ELK1, in the context of a c-fos promoter, stabilized SRF mutants, SRF154(A) and SRF153(A3), DNA binding, as revealed by the appearance of ternary complex factors (TCF). (E) Rescue of null SRF murine ES cells with lentiviral infections of SRFwt and SRF153(A3) was analyzed by heat maps of expressed genes generated on an Affymetrix array. Note that rescue with SRF153(A3) increased the expression of stem cell marker genes and cyclins but did not support the expression of many cardiac-specific genes and assembly genes needed for sarcomerogenesis.
Motif enrichment analysis of ATAC-seq peaks identified distinct sets of transcription factor binding sites in SRF vs. control, SRF153(A3) vs. control, and YAP5SA vs. control groups
| SRF vs. Control | SRF153(A3) vs. Control | YAP5SA vs. Control | ||||||
| Factor binding motif | E-value | Factor binding motif | E-value | Factor binding motif | E-value | |||
| ELF5 | 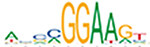 | 5.9e-296 | CTCF |  | 1.8e-290 | ETS1 |  | 1.7e-292 |
| ETS1 |  | 1.9e-201 | IRF3 |  | 2.0e-218 | SP2 |  | 1.8e-287 |
| ERG |  | 5.7e-195 | Fos:Jund |  | 4.1e-97 | ERG |  | 8.5e-283 |
| Fos:Jund |  | 1.4e-164 | ELF5 |  | 4.1e-94 | SP1 |  | 1.3e-253 |
| TEAD1 |  | 6.1e-138 | MEF2C |  | 8.7e-96 | JUNB |  | 1.1e-243 |
| FOS |  | 3.7e-137 | SP1 |  | 3.2e-84 | FOS |  | 2.3e-224 |
| SP1 |  | 3.3e-128 | RBPJ | 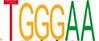 | 1.1e-69 | CTCF |  | 7.6e-157 |
| ELK3 | 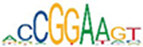 | 2.4e-127 | MEF2A |  | 1.6e-65 | IRF3 |  | 1.1e-136 |
| MEF2C |  | 6.8e-97 | RUNX1 |  | 8.3e-54 | RUNX1 |  | 1.5e-123 |
| CTCF | 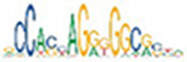 | 1.5e-65 | ETS1 |  | 6.5e-53 | MEF2C |  | 4.5e-96 |
| NFYB |  | 7.0e-55 | ERG |  | 1.1e-49 | SMAD3 |  | 5.5e-91 |
| RUNX3 |  | 3.7e-49 | GATA4 |  | 4.4e-44 | TEAD4 |  | 1.2e-76 |
| NFAT5 |  | 1.1e-37 | CREB1 |  | 1.2e-43 | TEAD1 |  | 1.1e-76 |
| STAT3 |  | 2.2e-25 | TEAD2 |  | 2.2e-40 | RBPJ |  | 5.9e-70 |
| SRF |  | 3.2e-15 | STAT3 |  | 7.4e-32 | SRF |  | 7.4e-29 |
| SRF |  | 6.4e-15 | SRF |  | 3.8e-4 | SRF |  | 2.6e-16 |
| GATA4 |  | 1.0e-7 | SRF |  | 2.1e-1 | GATA4 |  | 1.1e-13 |
RNA samples were hybridized against Affymetrix array 430a2 chips. Microarray raw data analysis was processed with dCHIP software (www.dchip.org). Microarray analysis confirmed the rescue of myogenic contractile protein gene activity with the lentiviral wild-type SRF expression [Figure 3E]. The ability for SRF to be the “myogenic driver” was completely abrogated in the SRF null ES cells. Rescue of SRF null ES cells with lentiviral expressed triplet SRF mutant, SRF153(A3), inhibited the induction of several cardiac myocyte specific genes, such as those encoding sarcomeric actins, heavy and light chain myosins, ion channels, and structural proteins. Expression of sarcomeric assembly factors such as ACTN2, NEBULIN, TITIN, MYOM1, OBSCN, and SMYD1 was suppressed in comparison to wildtype ES cells that formed cardiomyocytes following hanging drop formation. SRF mutant SRF153(A3) showed powerful activation of stem cell marker genes, such as Egr1, Rex1, Nanog, Oct4, Zic3, Dppa2, Dnmt1, Dnmt2, and proliferin, in comparison to SRF null ES cells and their rescue with wild-type SRF [Figure 3]. The expression of cyclins appeared to be repressed in the absence of SRF. Rescue with wild-type SRF caused activation of cyclins, CNNB1, CNND1, CNNC, and CNNE1, while SRF153(A3) strongly induced CNNA2, CNNB1, and CNNE1. Thus, SRF153(A3) elicited an imperfect or partial pluripotency program and activated the replication program, as evidenced by the appearance of cell cycle factors. The observation that a triple amino acid mutation in SRF153(A3) induced the expression of stem cell factors and blocked myocyte gene expression was unprecedented for switching gene regulatory programs.
Molecular modeling of the wild-type SRF and SRF153(A3) mutant roles in cardiac replication and differentiation is shown in Supplementary Figure 1. The diagrams show the inhibitory activity shared between myocyte replication and myocyte differentiation. The schematic diagram shows how phosphorylated ETS factors likely facilitate phosphorylated SRF binding to the c-fos promoter[5,11] to drive myocyte replication. Cofactors such as NKX2-5 and GATA4 expedite SRF binding to dependent cardiac actin gene promoters[8,9] to drive myocyte differentiation [Supplementary Figure 1A]. Conversely, the schematic model of SRF mutant SRF153(A3) supports cardiac myocyte replication because ELK1 stabilized SRF153(A3) binding to the c-fos promoter due to an adjacent ETS binding sequence (EBS). SRF153(A3) blocks cardiac differentiation because NKX2-5 and GATA4 cofactors fail to stabilize SRF(153A3) DNA binding to the sarcomeric cardiac actin promoter [Supplementary Figure 1B].
To exclude the potential low transfection rate of cardiomyocytes through plasmid DNA and the biosafety concerns of viral vectors, we applied a modified mRNA-based transfection system. Since mmRNA turns over, its expression is limited in contrast to viral vectors such as the adenovirus-associated virus, which express continually, blocking the recovery back to a contractile state. The strategy for generating base-modified synthetic mmRNA by T7 in vitro transcription is shown in Supplementary Figure 2A. A schematic diagram outlines our strategy to test transfection efficiency using GFP mmRNA at different ratios of RNA to Lipofectamine MessengerMAX. [Supplementary Figure 2B]. Approximately 45% of the tested NIH3T3 cells were transfected with the Lipo:mRNA ratio of 1.5 [Supplementary Figure 2C]. The validation of YAP5SA mmRNA expression activity by protein blots and nuclear detection is shown in Supplementary Figure 2D and E.
SRF153(A3) mmRNA transfection into neonatal rat ventricular myocytes induced stem cell and replication gene activity
Next, synthetic SRF153(A3) mmRNA transfection activity was evaluated in neonatal rat ventricular myocytes (NRVM) according to the protocol shown in Figure 4A. NRVM were transfected with SRF153(A3) mmRNA tipped with beta-globin 5’UTR. SRF153(A3) showed stronger expression than SRF153(A3) mmRNA without the beta-globin 5’UTR, as detected on a protein blot with an anti-SRF antibody [Figure 4B]. The relative amount of SRF153(A3) taken up into NRVM following a 6 h pulse and change of media showed a peak level (8500 units) at 12 h, which declined to about 85 units by 24 h and was undetectable by 48 h [Figure 4C], thus supporting the notion of the rapid turnover of mmRNA. Protein blots of extracts made from NRVM transfected with beta-globin 5’UTR-SRF153(A3) mmRNA for 24 h revealed the induction of NANOG by anti-NANOG staining [Figure 4D]. In further experiments, all synthetic mmRNA was made with beta-globin 5’UTR. As shown in Figure 4E, NRVM transfected with SRF153(A3) mmRNA presented a significant induction of NANOG and OCT4 but not KLF4 transcripts by quantitative PCR. Figure 4F shows NANOG expression by immunostaining. In addition, NANOG and OCT4 were upregulated within 24 h post-transfection, as assayed by RNA sequencing [Figure 4G]. In fact, in comparison to transfected YAP5SA, NANOG and OCT4 transcripts were induced to a greater extent with SRF153(A3), while YAP5SA upregulated KLF4 and N-MYC, and together they increased SOX2

Figure 4. SRF153(A3) mmRNA transfection into neonatal rat ventricular myocytes (NRVM) induced stem cell and replication gene activity. (A) Schematic diagram shows the synthetic mmRNA transfection protocol of NRVM. RNA was isolated from SRF153(A3) mmRNA plus Lipofectamine MessengerMAX treated and controlled Lipofectamine MessengerMAX only myocytes at time points that approximated the appearance of stem cell factors at 24 h, mitosis at 32 h, DNA packaging at 40 h, and the inhibition of cardiac specified genes at 40 h. (B) NRVM were transfected with SRF153(A3) mmRNA tipped at the 5’UTR end with or without beta-globin 5’UTR. SRF153(A3) was detected on a protein blot with an anti-SRF antibody. (C) NRVM were transfected with SRF153(A3) and, after 6 h, were washed with fresh media to remove excess RNA. Cellular RNA samples were collected at 12, 24, and 48 h, and the amount of SRF mRNA was evaluated by RT-PCR assay represented by closed circles and controls represented as open circles. (D) Protein blots of extracts from NRVM transfected with SRF153(A3) mmRNA assayed after 24 h revealed the appearance of NANOG by anti-NANOG staining. (E) NRVM transfected with SRF153(A3) mmRNA showed the significant induction of NANOG and OCT4 but not KLF4 transcripts by quantitative PCR compared to the control group, as determined by two-tailed statistical analysis in which *P < 0.05. (F) NRVM transfected with SRF153(A3) mmRNA showed immuno-staining with anti-NANOG was co-stained and merged with DAPI in comparison to control myocytes (lines are 10 µm in length). (G) Stem cell factors, NANOG, and OCT4 were upregulated in the SRF153(A3) mRNA treatment group within 24 h post-transfection assayed by RNA seq (Illumina) at 24 h post-transfection. (H) Increased expression of mitotic genes such as Bub1, Bub1b, Cenpe, Ndc80, CcnB1, and Dync1 was observed by 32 h post-SRF153(A3) treatment. (I) The appearance of DNA packaging genes, which mark the S phase of the cell cycle, occurred 40 h post-SRF153(A3) treatment, including Histone 1 genes such as Hist1h1a, Hist1h1b, and Hist1h2ba and the Histone 3 gene, Hist3h2ba. Other critical DNA packaging factor genes included, Npm1, Rbb4, ANP32B, Nap1|1, Hells, and SET. (J) At the height of the S phase by 40 h post-SRF153(A3) treatment, the inhibition of cardiac-specific gene expression was well on its way, including contractile proteins ACTC1, ACTN2A, and MYH6; transcription factors MYOCD and MEF2C; kinases AKAP12, MYLK3, AND GSK3B; and signaling factor VEGFA.
Combination of SRF153(A3) and YAP5SA mmRNA treated cardiomyocytes progress through the cell cycle
Next, we asked whether SRF153(A3) mmRNA complements YAP5SA mmRNA mutant activity to drive cardiomyocyte replication. Figure 5A illustrates the protocol we used to transfect NRVM with optimized solutions of SRF153(A3) mmRNA, YAP5SA mmRNA, or both together in combination with Lipofectamine MessengerMAX [Figure 5A]. NRVM have a very low replication rate of less than 1%-2% at the baseline. SRF153(A3) mmRNA was added to the NRVM once at the beginning of the first day with new media changes for 6 h. To identify replicating myocytes, 5-ethynyl-2′-deoxyuridine (alpha-EdU) was pulsed for 8 h to label any myocytes synthesizing DNA during the S phase of the cell cycle [Figure 5B]. Synthetic mRNA transfected myocytes were assessed for alpha-EdU incorporation. Virtually all the alpha-EdU+/DAPI stained nuclei lay within TNNT2-stained cells [Figure 5B]. Under confocal fluorescence microscopy, SRF153(A3) mmRNA induced DNA replication in 27% of the myocytes. YAP5SA mmRNA labeled 29% of the myocytes stained with EdU+/DAPI. Most exciting was the combination of synthetic mmRNA, which increased co-staining of the EdU+/DAPI marked myocytes to 35% of the total number of cells [Figure 5C and D]. This is likely an underestimate, since replicated cells may have synthesized DNA before the pulse of alpha-EdU. Nuclei co-stained with alpha-EdU (Red) and DAPI (Blue) were observed, as pink in the merged images. In addition, disorganized TNNT2 stained myofilaments (green) were observed with labeled alpha-EdU+/DAPI nuclei. Next, the ratio of alpha-EdU+-labeled cells tp nuclear stained DAPI was measured for NRVM pulsed at 24-32, 32-40, and 40-48 h with alpha-EdU [Figure 5E and F]. A significant drop in cells entering the S phase during the 32-40 and 40-48 h periods was observed in SRF153(A3) and combination groups, indicating transient gene delivery.

Figure 5. SRF153(A3) mutant and YAP5SA mutant mmRNA transduce NRVM proliferation. (A) Schematic protocol of transfected synthetic SRF153(A3) and YAP5SA mmRNA and/or in combination all with Lipofectamine MessengerMAX into NRVM to test for cell replication. (B) Optimal doses of synthetic mmRNA with Lipofectamine MessengerMAX were transfected in NRVM for 24 h and then pulsed for an additional 8 h with alpha-EdU. Controls did not receive mmRNA. NRVM were nuclear blue stained with DAPI. Alpha-EdU incorporated into the DNA of replicating myocytes was stained red with a Click-It kit. Anti-TNNT was used to stain organized contractile fibrils (green). White boxes mark replicated nuclei. (C) An increased number of myocytes is shown by the significant increase in DAPI stain nuclei for transfected SRF153(A3) and YAP5SA mmRNA as well as in combination. (D) EdU incorporation plotted as a ratio to DAPI was significantly increased in all mmRNA treatment groups compared to the control group, as determined by two-tailed statistical analysis, in which *P < 0.05 and **P < .005. (E) The ratio of EdU/DAPI-labeled cardiomyocytes was quantitated at three different time points, each with a final 8 h EdU pulse. (F) The first 24 h revealed the highest ratio of alpha-EdU/DAPI-labeled cardiomyocytes for the combination of mmRNA. The following time points showed reduced alpha-EdU/DAPI ratios, indicating declining EdU incorporation per nucleus. The number of cells ranged from 700 to 2800 in triplicates.
Would a combination of SRF153(A3) and YAP5SA mmRNA induce proliferation in adult cardiomyocytes? The experimental protocol, as shown in Figure 6A, used mouse adult cardiomyocytes isolated from the left ventricle of 12-week-old mice by the Langendorff system. Anti-TUB was used to show the rectangular appearance of adult myocytes. The Lipofectamine MessengerMax control, wild-type SRF, and YAP-treated groups barely displayed any alpha-EdU incorporation [Figure 6B]. Both SRF153(A3) and YAP5SA treatments strongly induced alpha-EdU incorporation [Figure 6C], while, notably, more than 90% of cardiomyocytes had an alpha-EdU positive signal in the co-transfected combination group [Figure 6D]. Aside from inducing adult myocyte nuclear replication, we noticed that the nuclei in the SRF153(A3) and co-treatment group, as shown in Figure 6E and F, are mixtures of bi- and tetraploid nuclei. In addition, DIAPH3 was localized to multiple regions between and surrounding dividing nuclei (shown by white arrows in Figure 6E). DIAPH3 marks anaphase of the cell cycle and induces F-actin to help assemble a contractile ring during cytokinesis. In addition, anti-TNNT-stained adult mouse cardiomyocytes

Figure 6. Transfected SRF153(A3) and YAP5SA mmRNA induced nuclear replication and the appearance of cleavage furrows in adult rat cardiomyocytes. (A) Schematic diagram of SRF153(A3) and YAP5SA mmRNA mutants and/or in combination transfected into three-month-old rat primary cardiac myocytes isolated by the Langendorff system. Alpha-EdU was pulsed in cultures to stain DNA synthesis in nuclei within 24 h after mmRNA transfection. (B) Alpha-EdU pulse assay of adult CM treated with wild-type SRF and YAP. Control myocytes were treated with Lipofectamine MessengerMAX. Wild-type SRF and the YAP-treated group barely displayed the alpha-EdU signal. (C) Alpha-EdU pulse assay of adult CM treated with SRF153(A3) and YAP5SA. (D) Notably, more than 90% of cardiomyocytes had alpha-EdU/nuclei positive signals, which was significantly increased in all mmRNA treatment groups compared to the control group, as determined by two-tailed statistical analysis, in which *P < 0.05. (E) Adult CM treated SRF153(A3) and YAP5SA were stained with DAPI, anti-TNNT, and anti-DIAPH3. Immuno-fluorescence microscopy showed DIAPH3 localized in multiple regions between and surrounding dividing nuclei, as shown by white arrows. (F) TNNT marker in the mRNA-treated adult mouse cardiomyocytes showed reduced TNNT immunofluorescence around the nuclei (marked by dashed circles) in the SRF153(A3) mmRNA and the SRF153(A3) and YAP5SA mmRNA combo treated group. Cell cycle cleavage furrows were marked by white arrows.
Combination of SRF153(A3) and YAP5SA mmRNA treated cardiomyocytes show a proliferative gene profile
We performed next-generation RNA sequencing (RNA-seq) on SRF153(A3)- and/or YAP5SA-treated NRVM using the protocol shown in Figure 7A. Compared to the control, more than 2000 DEG were recognized in the combination-treated group, with p- Adj-value (FDR correction) < 0.1 and log2 fold change > 1 or < -1. Among the top 20 regulated genes, SRF and YAP1 were increased 11- and 64-fold in the combination-treated group, with the two lowest P-values, respectively, indicating that our mmRNA-based overexpression system is effective in myocytes [Supplementary Figure 4]. Graphical presentations of the principal component analysis (PCA) plot the distance of samples under Ctrl, SRF153(A3), YAP5SA, and combination treatment based on the normalized expression values using DESeq2. The PCA plots demonstrated well-separated clusters at all three time points based on the level of gene expression

Figure 7. Cardiomyocytes transfected with a combination of SRF153(A3) and YAP5SA mmRNA mutants showed a primitive genetic expression profile. (A) Schematic diagram of NRVM transfected with SRF153(A3) and YAP5SA mmRNA mutants individually and in combination with optimized Lipofectamine MessengerMAX. RNA was collected for RNA sequencing. (B) Heat map of cytokinesis (GO:0000910) genes generated from RNA-seq data at 32 h after transfection. (C) Heat map of spindle assembly (GO: 0051225) genes generated from RNA-seq data 40 h after transfection. (D) Heat map of cardiac muscle cell differentiation (GO:0055007) genes generated from RNA-seq data 48 h after transfection. (E) GSEA plot for differentially regulated genes for DNA replication and cell cycle [NES (normalized enrichment score, relative to control) and FDR q-value (after FDR correction)]. (F) GO-term analysis of upregulated DEG genes in 48 h post-transfection combination group versus the control group (top). GO-term analysis of downregulated DEG genes in 48 h post-transfection combination group versus the control group (bottom).
We generated several heat maps to identify specific upregulated and downregulated genes [Figure 7]. A map of cytokinesis (GO:0000910) genes was generated from RNA-seq data at 32 h after transfection [Figure 7B]. For example, genes required for cytokinesis such as PLK1 (Polo-like kinase 1) promote contractile ring formation and cleavage furrow ingression and foster RhoA accumulation at the equator[34]. Anillin (ANLN) was also highly upregulated in the combination treatment group. The presence of ANLN is required in multiple stages of cytokinesis, and ANLN acts as a key mediator of cytokinesis[35]. Mitotic spindle forms during cell division and separates sister chromatids between daughter cells[36]. RNA-seq data show that several spindle assembly factors (GO: 0051225), such as BUB1 (mitotic checkpoint serine/threonine-protein kinase BUB1), were activated after synthetic mRNA transfection [Figure 7C and Supplementary Figure 6]. Cardiac-specific genes were downregulated after mmRNA treatment. SRF153(A3) alone can decrease the expression level of certain cardiac-specific genes, but, in combination with YAP5SA, cardiac differentiation factors were strongly repressed [Figure 7D and Supplementary Figure 7]. Genes involved in cardiac muscle cell differentiation (GO:0055007) were downregulated in the combination treatment group [Figure 7D]. ACTN2 (actinin alpha 2) encodes a muscle-specific alpha-actinin isoform expressed in cardiac muscles and skeletal muscles. MYH6 (myosin heavy chain 6), one of the two alpha heavy chain subunits of cardiac myosin, crucial genes contributing to cardiac muscle cell differentiation, was inhibited, suggesting that co-transfectant SRF153(A3) and YAP5SA mmRNA pushed cardiomyocytes into a less differentiated, more primitive stage.
GSEA is a computational method that determines whether a priori defined set of genes shows statistically significant and concordant differences between two groups[28]. Here, the GSEA analysis showed robust upregulation of genes in the “KEGG cell cycle” as well as “KEGG DNA replication” gene sets in the combination treatment group compared to the control [Figure 7E]. The normalized enrichment score (NES) of both enrichment analyses reached more than 1.9 with a q-value less than 0.01, indicating an extreme enrichment for these upregulated genes in the two gene sets. Overall, these results indicate that combination treatment of SRF153(A3) and YAP5SA induces cell replication by increasing cell cycle regulation gene expression and decreasing the cardiomyocyte function gene expression. The combination of SRF153(A3) and YAP5SA synthetic mRNA transfection promotes cardiomyocyte DNA replication.
We next performed gene ontology (GO) term analysis on both upregulated and downregulated DEG genes in the 48 h post-transfection combination treatment group versus the control group [Figure 7F, top]. We found that “cell division”, “regulation of mitotic cell cycle”, and “nuclear division” terms were significantly enriched in terms of biological process (BP) in the upregulated DEG enrichment analysis. In contrast, the downregulated DEG is specifically enriched in “muscle tissue development” and “muscle system process” in terms of BP and “contractile fiber” and “myofibril” in terms of CC [Figure 7F, bottom].
Since GSEA identified upregulation of the genes involved in DNA replication, we reviewed the RNA-seq data at the earliest time points and drew attention to the genes that contribute to the DNA replication process and replisome. ORC2 (origin recognition complex subunit 2), which recruits NOC3 (nucleolar complex-associated protein 3), CDC6 (cell division cycle 6), CDT1 (chromatin licensing and DNA replication factor 1), and MCM2-7 (minichromosome maintenance complex component 2-7) in the pre-initiation phase in early G1 stage were robustly upregulated by the combination mRNA treatment [Figure 8A]. A model of the core eukaryotic DNA replication is shown in Figure 8B. In the initiation stage, CDC45, which is essential for both the initiation and elongation stages of DNA replication, showed upregulation. After the MCM2-7 complex formation, CDC45 assembles onto the origin in the late G1 stage and concurrent with the onset of initiation. In the elongation stage, for example, CLASPIN was highly upregulated by the combination treatment. CLASPIN is a component of the replisome and contains a domain for docking with CHK1 (checkpoint kinase). CLASPIN promotes checkpoint signaling at the replisome. Most of the crucial genes involved in DNA replication, such as ORC2, MCM2, CDC45, and CLASPIN, were significantly increased in the combination treatment group [Figure 8A and B], suggesting that the SRF153(A3) and YAP5SA combination mmRNA treatment promote cardiomyocyte proliferation by fostering cell cycle entry by promoting DNA replication in the G1 phase. Upregulated genes under combination treatment are shown in red in Figure 8B. Thus, SRF153(A3) and YAP5SA introduction in cardiomyocytes are sufficient to promote cardiomyocyte proliferation by pushing every stage, from the initiation of DNA replication to the beginning of cytokinesis.

Figure 8. Cardiomyocytes transfected with a combination of SRF153(A3) and YAP5SA mmRNA mutants induced DNA replication and replisome genes. (A) Heat map of DNA replication and replisome genes at 32 h post-transfection between combination treatment group and control group. (B) Model of the core eukaryotic DNA replication. Upregulated genes under SRF153(A3) and YAP5SA combination treatment are shown in red.
SRF153(A3) and YAP5SA mmRNA increased cardiomyocyte chromatin accessibility
To test if chromatin remodeling occurs in response to our synthetic mmRNA treatment and to identify downstream gene targets, we performed ATAC-sequencing 24 h post-transfection. The PCA plot is shown in Figure 9A, which suggests that different samples were separated well according to their peak annotation results. Using untreated groups as a reference, we identified 5610, 15,571, 7087, 20,217, and 12,156 unique peaks in SRF, SRF153(A3), YAP, YAP5SA, and combination treatment groups, respectively. These results suggest a substantial increase of accessible chromatin region in the SRF153(A3) and YAP5SA treatment group compared to the untreated control and wildtype controls. Unique peaks within the ±2 kb regions surrounding the TSS of 2842, 2638, and 1506 genes were annotated in the promoter region in the SRF153(A3), YAP5SA, and combination group, respectively, indicating a large portion of the peaks were potentially related to the transcriptional activities [Figure 9B]. Genes involved in nuclear division had more accessible promotor regions [Figure 9C] in the SRF153(A3), YAP5SA, and combination treatment groups, suggesting that our mRNA treatment pushed multiple stages of the cardiomyocyte cell cycle. Heat maps of DNA replication (GO:0006260) genes and nuclear division (GO: 0000280) genes were plotted using ATAC-seq signal intensity score using the peak signal within the ±2 kb regions surrounding the TSS in each sample ATAC-seq signal intensity score [Supplementary Figure 8]. Enhanced signals at the promoters of marker genes involved in positive regulation of cell cycle (GO:0045787) and spindle assembly (GO:0051225) were observed in SRF153(A3), YAP5SA, and combination treatment groups [Figure 9C], indicating that the mRNA treatments fostered accessible promoter regions of these critical genes involved in multiple steps of the cell cycle, thus fostering cell replication gene restructuring activity. We performed the GO analysis in terms of biological processes on the combination of unique peaks [Figure 9D]. Negative regulation of programmed cell death (GO: 0043069), regulation of cell proliferation (GO:0042127), and negative regulation of apoptotic process (GO: 0053066) were identified as the top enriched terms for the combination treatment group compared to blank control. Heat maps plotted using ATAC-seq signal intensity score showed that many genes involved in DNA replication (GO:0006260), nuclear division (GO:0000280), spindle assembly (GO: 0051225), and positive regulation of cell cycle (GO:0045787) had increased peak signals in SRF153(A3), YAP5SA, and combination treatment groups compared to blank control and wildtype controls [Figure 9C and Supplementary Figures 8 and 9].

Figure 9. SRF153(A3) and YAP5SA mmRNA treated cardiomyocytes induced chromatin accessibility. (A) Principal component analysis (PCA) plot of ATAC-seq data. (B) Heat map showing genome-wide chromatin accessibility using the read depth. (C) Heat maps of ATAC-seq signal intensity of the genes involved in DNA replication (GO:0006260), nuclear division (GO: 0000280), and positive regulation of cell cycle (GO:0045787). (D) Gene ontology analysis in terms of biological processes on the combination of unique peaks.
To identify the underlying mechanism of how SRF153(A3) works as a novel transcription factor, we used ATAC-sequencing to create a bioinformatics landscape of interactomes of SRF153(A3), SRF, and YAP5SA [Table 1]. Wild-type SRF DNA binding targets revealed strong preferences for interaction with ETS factors with the highest E-values of Elf5 (5.9e-296), ETS1 (1.9e-201), and ERG (5.7e-195). In addition, strong interactions were shown for TEAD1 (6.08e-138), but there were much weaker interactions with SRF’s CArG boxes at 3.23e-15, demonstrating natural weaker binding for SRF, as previously shown[10]. SRF153(A3)’s interactome showed a broader range of transfer factor interactions, such as strong preference for CTCF (1.8e-290) and SP1 (3.2e-84), both C2H2 zinc finger factors; RBPJ (1.1e-69) and a Rel homology region cofactor; several ETS factors, ELF5 (4.1e-94) and ETS1(6.5e-53); TEAD2 (2.2e-40) factors; and GATA4 (4.4e-44). As shown in Figure 2 and Table 1, alanine substitution mutations in N-terminal extension of the MADS box for 154 lysine or SRF153(A3) severely weakened DNA binding activity to CArG boxes (3.8e-4 and 5.2e-1). Thus, SRF153(A3) diminished the transcription of many CArG-dependent cardiac-specified genes, assuming the formation of SRF153(A3) homodimers and SRF153(A3):SRF heterodimers. In addition, numerous cofactors recruited SRF153(A3) to bind other DNA sequences, typically not associated with SRF, such as IRF3 (2.0e-218), FOS:JUND (4.1e-97), MEF2C (8.7e-96), RUNX1 (8.3e-54), and STAT3 (7.4e-32). YAP protein interacts with myriad transcription factors, yet only a few showed interactions with the YAP phosphorylation mutant, such as the TEADSs, EGR1, TBX5, SMADS, and RUNX. Since YAP5SA does not directly bind to DNA, ATAC-seq also revealed novel interactomes with DNA binding cofactors. YAP5SA is likely recruited to ETS1 (1.7e-292), SP2 (1.8e-287), SP1 (1.3e-253), JUNB (1.1e-243), FOS (2.3e-224), CTGF (7.6e-157), IRF3 (1.1e-136), MEF2C (4.5e-96), and RBPJ (5.9e-70), as well as RUNX1 (1.5e-123), SMAD3 (5.5e-91), and TEAD1 (1.1e-76). YAP5SA target sequencing also revealed considerable interactions with SRF (7.4e-29), previously not shown. Therefore, many cofactors that co-interact with SRF153(A3) and YAP5SA are similar in binding specificity if not identical, thus augmenting the spectrum of gene targets, many of which support cellular replication and growth.
Another important feature revealed by ATAC-seq was the chromatin remodeling of many growth factors and signaling pathways usually active during early embryo development and cardiogenesis. Supplementary Figure 10A shows gene families representing FGFs, BMPs, Notchs, and Wnts having their chromatin remodeled during a 24 h cellular treatment with SRF153(A3) and YAP5SA mmRNA. Many of these genes were remodeled by YAP5SA treatment. NOTCH2 can modify the transcriptome and cellular electrophysiology of cardiomyocytes to resemble cells of the specialized conduction system and not the differentiated contractile phenotype[37]. Several of these factors, such as the non-canonical WNT5A/B and WNT11, augment cardiomyogenic proliferation and differentiation of human cardiac progenitor cells[38]. WNT5A and WNT11 also regulate second heart field development in mice, and their genetic deletion causes the loss of second heart field progenitors[39]. WNT5A/WNT11 inhibits CTNNB1 signaling and promotes cardiac progenitor development in differentiating embryonic stem cells[39]. Signaling pathways that express STATS and JAKS, such as STAT5 and JAK3, have key roles in cellular growth, apoptosis, and transformation[40]. JAK3 is expressed in immune cells and transduces a signal in response to its activation via tyrosine phosphorylation by interleukin receptors. In addition, YAP signaling has a strong impact on inducing IGF1, IGF2, and their binding proteins and gene remodeling [Supplementary Figure 10A] to enhance cell growth and resist apoptosis[41].
The lack of telomerase activity is a barrier to cardiomyocyte proliferation and cardiac regeneration[42]. Most remarkable was the restructuring of many genes associated with the GO term for telomere maintenance facilitated by SRF153(A3) [Supplementary Figure 10B]. Telomerase reverse transcriptase 1(TERT1) and a member of the shelterin complex, ACD or TPP1, which facilitate the replication of the double-stranded telomeric DNA tracts and protect the telomeric end from unregulated DNA repair, showed enhanced chromatin remodeling by SRF153(A3)[43]. TCP1 (t-complex protein 1) causes positive regulation of telomere maintenance via telomerase. TCP1 and CCT7 are members of the chaperonin containing TCP1 complex (CCT), also known as the TCP1 ring complex[36]. In addition to TERT, genes encoding Tep1, NAF1, STN1, and RTEL1, associated with roles in cardiac telomeric myopathies, displayed chromatin restructuring[43]. STN1 contributes to telomere maintenance CST protection of telomeres under conditions of replication stress. Telomere maintenance GO analysis also showed genes associated with the recombination-dependent alternative lengthening of telomeres (ALT) pathway, a pathway independent of TERT that involves DNA recombination[44]. Nevertheless, activation of a telomere maintenance mechanism (TMM) or ALT pathway may play vital roles in lengthening shortened telomeres and the rejuvenation of cardiac myocytes[45].
DISCUSSION
The prevalence of myocardial insufficiency in diseased hearts underscores the reality that the intrinsic regenerative capacity of the adult heart is insufficient to repair a substantive injury[1]. Considerable effort has therefore been invested in developing interventions aimed at gaining functional cardiomyocytes and enhancing the function of surviving cardiomyocytes in diseased hearts. Herein, we describe a combination of factors that effectively induce cardiomyocyte proliferation in vitro. The SRF mutant, SRF153(A3), blocked cardiac differentiation in culture, yet it propelled cardiomyocytes to express stem and cell cycle gene activity. SRF153(A3) shares similar attributes to the YAP mutant, YAP5SA, a HIPPO signaling factor that promotes cell growth. Our strategy was to generate synthetic modified SRF153(A3) and YAP5SA mmRNA for highly efficient transfection into cardiomyocytes. We observed robust expression of nuclear DNA replication fork and replisome pathway genes, the center point of initiating DNA replication. Bioinformatics analysis revealed the upregulation of multiple cell cycle gene clusters with co-expression of SRF153(A3) and YAP5SA, while gene clusters associated with cardiomyocyte differentiation (GO: 0055007), sarcomeric assembly, and cardiac muscle contraction (GO: 0060048) were profoundly downregulated. SRF153(A3) and YAP5SA promoted cardiomyocyte proliferation by inhibiting SRF-dependent cardiomyocyte differentiation, thus pushing cardiomyocytes into a more primitive stage to foster cell replication.
Support for this idea was underscored by increased chromatin accessibility of embryonic morphogenetic factors, such as WNT5A, WNT5B, and WNT11, BMP, FGF, and NOTCH signaling factors, all of which are important for embryonic cardiac myocyte proliferation and patterning. Non-canonical WNT signaling through WNT5A/B and WNT11 genes regulates cell migration and the transition of cardiac progenitors to differentiated cardiomyocytes. Under NOTCH2 signaling, cardiomyocyte differentiation is directed towards conduction-like cell types[37], while BMP2 signaling is essential for cardiac cushion formation, the epithelial-mesenchymal transition, and myocardial patterning[46]. FGF8 is needed for anterior heart field development[47]. YAP may direct cardiomyocyte proliferation and heart size in part through insulin-like growth factor signaling[41]. The increased remodeling of IGF1 and IGF2 genes and their associated binding protein genes replicate an observation made for the expression of these potent growth factors and survival factors following YAP mutant treatment of myocytes[41].
Telomere shortening is a hallmark of the aging process leading to cellular senescence[42,44]. Telomerase activity is limited to the early stages of embryonic development and adult stem cells[36]. The appearance of remodeled genes for telomere maintenance is another feature supporting the notion of returning to a more embryonic-like state. SRF153(A3) was responsible for altering the TERT gene structure. In adult tissues, telomere extension is through recombination independent from TERT, likely by ALT[42,44,45]. We observed ATAC-seq structural changes driven by SRF153(A3) for SLX1, an important regulator of t-circles, and as a catalytic subunit with SLX4, it is an endonuclease that resolves DNA structures formed during repair and recombination[48]. In addition, structural changes were increased for genes encoding ATM, a master controller of cell cycle checkpoint pathways, which responds to DNA damage and HSP90AA1, another telomere maintenance factor. Whether telomere extension could also be through DNA recombination at telomeres, SRF153(A3) remodeling of TERT is novel and consistent with the overall return of myocytes to a highly proliferative state.
Our findings suggest a complementary effect of YAP5SA and SRF153(A3) in promoting cardiomyocyte replication. In some cases, YAP5SA and SRF153(A3) synergized, while we observed additive effects in regulating cell cycle gene expression and alpha-EdU incorporation in cardiomyocytes. However, YAP is not a direct cofactor of SRF. SRF has several tissue-specific regulatory cofactors that control SRF activity by interacting with SRF’s MADS box, whereas YAP, as the transcription co-activator of the HIPPO signaling pathway, does not directly bind to DNA. TEAD1 or TEF1, one of SRF’s cofactors shown by our previous studies[20,21] to physically interact with SRF, may also serve as a bridge between YAP5SA and SRF153(A3) to implement the synergy between the combination factors. In fact, analysis of SRF153(A3) binding targets by ATAC sequencing showed significant enrichment for binding to the mammalian TEAD family of transcription factors, which contain four members, TEAD1, TEAD2, TEAD3, and TEAD4. The TEA/ATTS domain of TEAD1 interacts with the MADS box of SRF, and these two transcription factors were reported to co-activate the skeletal α-actin promoter in COS-1 cells[21]. Transcription factor TEAD1 bound to the TEAD-binding domain of YAP/TAZ may serve as the main binding platform of YAP/TAZ.44 The interaction between YAP1 and TEAD1 is required to stimulate cardiomyocyte proliferation[49]. Therefore, given the significant role of TEAD1 in both SRF-TEAD1 and YAP-TEAD1 complexes, it is suggested that the recruitment of TEAD1 may be necessary for SRF153(A3)-YAP5SA interaction. However, identifying the underlying mechanism of how SRF153(A3) works as a novel synthetic transcription factor, we generated interactomes for SRF153(A3), SRF, and YAP5SA [Table 1]. Many transcription factors that facilitate SRF153(A3) recruitment to the many sites other than CArG boxes were noted. Similar to wild-type SRF, SRF153(A3) also prefers recruitment by ETS factors, CTCF, SP1, RBPJ, NFAT5, and TEAD1. However, the mutation of 154 lysine in the MADS box severely weakened DNA binding activity to CArG boxes. Thus, SRF153(A3) diminished the transcription of many CArG-dependent cardiac-specified genes. In addition, we found many new YAP5SA cofactor interactomes with DNA binding cofactors ETS1, SP2, SP1, JunB, Fos, CTGF, IRF3, Mef2C, and RBPJ, as well as its well-known cofactors RUNX1, SMAD3, and TEAD1. YAP5SA target sequencing also revealed considerable interactions with SRF and its cofactors, previously not shown. Thus, SRF153(A3) and YAP5SA share interactomes with many more transcription factors than previously thought, providing a powerful spectrum of transcription regulators that are strongly pro-replicative.
Synthetic mRNA may be used as a safe and efficient gene delivery vehicle in adult hearts. Compared to viral vectors, the transient gene expression that mRNA provides is far more controllable, which makes the mmRNA gene-delivery method a safer option for delivering therapeutic factors for cardiac regeneration. Given the post-transcription nature of mRNA, mmRNA does not require transfer to the nucleus to get the expression of the target protein. Besides, mmRNA-based gene delivery can deliver gene combinations with different ratios specifically tailored to patients with a different course of the disease. Our data suggest that synthetic mRNA may be used to deliver SRF153(A3) and YAP5SA into adult cardiomyocytes both in vitro and in vivo to achieve high transfection efficiency with little biosafety concern.
DECLARATIONS
Authors’ contributions
Performed many of the molecular biology experiments and drafted the manuscript: Xiao S
Performed part of the molecular biology experiments and edited the manuscript: Liang R, Muili AB
Performed bioinformatics analysis of the data and edited the manuscript: Cao X
Designed part of the experiments, interpret the data, and edited the manuscript: Navran S
Developed the concept, designed part of the experiments, interpreted the data, and wrote the manuscript: Schwartz RJ
Designed experiments, interpreted the data, and edited the manuscript: Iyer D
Availability of data and materials
RNA-Seq and ATAC-Seq data will be submitted to GEO and will be available to the public upon release.
Financial support and sponsorship
Research support for this study to RJS was through the University of Houston, a Cullen Endowed Chair, Texas Board of Higher Education, Leducq Foundation and a Sponsored Research Agreement from Animatus Biosciences, LLC. The funders had no role in the preparation of the manuscript or decision to publish this manuscript.
Conflicts of interest
Navran S, Schwartz RJ, and Iyer D are founders of Animatus Biosciences. Other authors, Xiao S, Liang R, Muili AB, and Cao X declared that there are no conflicts of interest. This research was underwritten in part by a grant from Animatus Biosciences LLC, in which there is a financial interest. A management plan, reviewed and recommended by the University of Houston Conflict of Interest Committee as acceptable to address the conflict.
Ethical approval and consent to participate
C57BL/6J of both genders were housed and studied in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (eighth edition)[31]. Animals were handled according to Institutional Animal Care and Use Committee (IACUC) protocols 15-055 and 16-015 (University of Houston).
Consent for publication
Not applicable.
Copyright
© The Author(s) 2022.
Supplementary Materials
REFERENCES
1. Porrello ER, Mahmoud AI, Simpson E, et al. Transient regenerative potential of the neonatal mouse heart. Science 2011;331:1078-80.
2. Foglia MJ, Poss KD. Building and re-building the heart by cardiomyocyte proliferation. Development 2016;143:729-740.
3. Ocampo A, Reddy P, Martinez-Redondo P, et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 2016;167:1719-33.
4. Chen Y, Lüttmann FF, Schoger E, et al. Reversible reprogramming of cardiomyocytes to a fetal state drives heart regeneration in mice. Science 2021;373:1537-40.
5. Treisman R. Ternary complex factors: growth factor regulated transcriptional activators. Curr Opin Genet Dev 1994;4:96-101.
6. Miano JM. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol 2003;35:577-93.
7. Pellegrini L, Tan S, Richmond TJ. Structure of serum response factor core bound to DNA. Nature 1995;376:490-8.
8. Chen CY, Schwartz RJ. Recruitment of the tinman homolog Nkx-2.5 by serum response factor activates cardiac alpha-actin gene transcription. Mol Cell Biol 1996;16:6372-84.
9. Sepulveda JL, Vlahopoulos S, Iyer D, Belaguli N, Schwartz RJ. Combinatorial expression of GATA4, Nkx2-5, and serum response factor directs early cardiac gene activity. J Biol Chem 2002;277:25775-82.
10. Niu Z, Iyer D, Conway SJ, et al. Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc Natl Acad Sci 2008;105:17824-9.
11. Janknecht R, Nordheim A. Elk-1 protein domains required for direct and SRF-assisted DNA-binding. Nuclei Acids Res 1992;20:3317-24.
12. Murai K, Treisman R. Interaction of serum response factor (SRF) with the Elk-1 B box inhibits RhoA-actin signaling to SRF and potentiates transcriptional activation by Elk-1. Mol Cell Biol 2002;22:7083-92.
13. Wang DZ, Li S, Hockemeyer D, et al. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc Natl Acad Sci U S A 2002;99:14855-60.
14. Chang DF, Belaguli NS, Iyer D, et al. Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Dev Cell 2003;4:107-18.
15. Vartiainen MK, Guettler S, Larijani B, Treisman R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Scienc 2007;316:1749-52.
16. Lockman K, Hinson JS, Medlin MD, Morris D, Taylor JM, Mack CP. Sphingosine 1-phosphate stimulates smooth muscle cell differentiation and proliferation by activating separate serum response factor co-factors. J Biol Chem 2004;279:42422-30.
17. Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003;113:329-42.
18. Yu OM, Miyamoto S, Brown JH. Myocardin-related transcription factor A and yes-associated protein exert dual control in G protein-coupled receptor- and rhoa-mediated transcriptional regulation and cell proliferation. Mol Cell Biol 2015;36:39-49.
19. Zaromytidou AI, Miralles F, Treisman R. MAL and ternary complex factor use different mechanisms to contact a common surface on the serum response factor DNA-binding domain. Mol Cell Bio 2006;26:413448.
20. Gupta M, Kogut P, Davis FJ, Belaguli NS, Schwartz RJ, Gupta MP. Physical interaction between the MADS box of serum response factor and the TEA/ATTS DNA-binding domain of transcription enhancer factor-1. J Biol Chem 2001;276:10413-22.
21. MacLellan WR, Lee TC, Schwartz RJ, Schneider MD. Transforming growth factor-beta response elements of the skeletal alpha-actin gene. Combinatorial action of serum response factor, YY1, and the SV40 enhancer-binding protein, TEF-1. J Biol Chem 1994;269:16754-60.
22. Xiao Y, Leach J, Wang J, Martin JF. Hippo/Yap signaling in cardiac development and regeneration. Curr Treat Options Cardiovasc Med 2016;18:38.
23. Del Re DP, Yang Y, Nakano N, et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem 2013;288:3977-88.
24. Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 2007;21:2747-61.
25. Foster CT, Gualdrini F, Treisman R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev 2017;31:2361-75.
26. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 2016;34:5257.
27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550.
28. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545-50.
29. Wu T, Hu E, Xu S, et al. ClusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (Camb) 2021;2:100141.
30. Kolde R. Pheatmap: pretty heatmaps - R package version. Available from: https://rdrr.io/cran/pheatmap/ [Last accessed on 16 May 2022].
31. National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the care and use of laboratory animals. 8th edition. Washington (DC): National Academies Press (US); 2011.
32. Schratt G, Philippar U, Berger J, Schwarz H, Heidenreich O, Nordheim A. Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J Cell Biol 2002;156:737-50.
33. Pires DEV, Ascher DB. MCSM-NA: predicting the effects of mutations on protein-nucleic acids interactions. Nucleic Acids Res 2017;45:W241-W246.
34. Petronczki M, Glotzer M, Kraut N, Peters JM. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev Cell 2007;12:713-25.
35. Zhang S, Nguyen LH, Zhou K, et al. Knockdown of anillin actin binding protein blocks cytokinesis in hepatocytes and reduces liver tumor development in mice without affecting regeneration. Gastroenterology 2018;154:1421-34.
36. Walczak CE, Heald R. Mechanisms of mitotic spindle assembly and function. Int Rev Cytol 2008;265:111-58.
37. Rentschler S, Yen AH, Lu J, et al. Myocardial notch signaling reprograms cardiomyocytes to a conduction-like phenotype. Circulation 2012;126:1058-66.
38. Pandur P, Läsche M, Eisenberg LM, Kühl M. Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature 2002;418:636-41.
39. Cohen ED, Miller MF, Wang Z, Moon RT, Morrisey EE. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development 2012;139:1931-40.
40. Leonard WJ. Role of Jak kinases and STATs in cytokine signal transduction. Int J Hematol 2001;73:271-7.
41. Xin M, Kim Y, Sutherland LB, et al. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal 2011;4:ra70.
42. Aix E, Gallinat A, Flores I. Telomeres and telomerase in heart regeneration. Differentiation 2018;100:26-30.
43. Henslee G, Williams CL, Liu P, Bertuch AA. Identification and characterization of novel ACD variants: modulation of TPP1 protein level offsets the impact of germline loss-of-function variants on telomere length. Cold Spring Harb Mol Case Stud 2021;7:a005454.
44. Madrid A, Rode L, Nordestgaard BG, Bojesen SE. Short telomere length and Ischemic heart disease: observational and genetic studies in 290 022 individuals. Clin Chem 2016;62:1140-9.
45. Zhang JM, Zou L. Alternative lengthening of telomeres:from molecular mechanisms to therapeutic outlooks. Cell Biosci 2020;10:30.
46. Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005;132:5601-11.
47. Ilagan R, Abu-Issa R, Brown D, et al. Fgf8 is required for anterior heart field development. Development 2006;133:2435-45.
48. Wilson JS, Tejera AM, Castor D, Toth R, Blasco MA, Rouse J. Localization-dependent and -independent roles of SLX4 in regulating telomeres. Cell Rep 2013;4:853-60.
Cite This Article
How to Cite
Xiao, S.; Liang R.; Muili A. B.; Cao X.; Navran S.; Schwartz R. J.; Iyer D. Mutant SRF and YAP synthetic modified mRNAs drive cardiomyocyte nuclear replication. J. Cardiovasc. Aging. 2022, 2, 29. http://dx.doi.org/10.20517/jca.2022.17
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.